Differential Accessibility in ATAC-seq
Learning outcomes
to perform GC-aware normalisation of ATAC-seq data using
EDASeqto detect differentially accessible regions using
edgeR
Introduction
In this tutorial we use an R / Bioconductor packages EDAseq and edgeR to perform normalisation and analysis of differential accessibility in ATAC-seq data.
Data & Methods
We will build upon the main lab ATACseq data analysis:
first we will summarise reads to detected peaks using subset data as an example to obtain a counts table;
we will use the counts table encompassing complete data for differential accessibility analysis;
Setting-up
We need access to file nk_merged_peaks.saf which we created in the ATACseq data analysis, part “Merged Peaks” and bam files with alignments.
Assuming we start at atacseq/analysis:
mkdir counts_table
cd counts_table
ln -s ../peaks/consensus/nk_merged_peaks.saf
ln -s ../../data_proc/* .
If you haven’t followed the peak calling and merging lab, you can continue from this point by linking necessary files:
ln -s ../../results/peaks/consensus/nk_merged_peaks.saf
ln -s ../../data_proc/* .
Data Summarisation
We can now summarise the reads allowing for 20% overlap of the read length with peak feature (--fracOverlap 0.2) and counting fragments rather than reads (-p for PE):
module load subread/2.0.0
featureCounts -p -F SAF -a nk_merged_peaks.saf --fracOverlap 0.2 -o nk_merged_peaks_macs3.counts ENCFF363HBZ.chr14.proc.bam ENCFF398QLV.chr14.proc.bam ENCFF828ZPN.chr14.proc.bam ENCFF045OAB.chr14.proc.bam
Let’s take a look inside the counts table using head nk_merged_peaks_macs3.counts.
nk_merged_peaks_macs3.counts
head nk_merged_peaks_macs3.counts
# Program:featureCounts v2.0.0; Command:"featureCounts" "-p" "-F" "SAF" "-a" "nk_merged_peaks.saf" "--fracOverlap" "0.2" "-o" "nk_merged_peaks_macs3.counts" "ENCFF363HBZ.chr14.proc.bam" "ENCFF398QLV.chr14.proc.bam" "ENCFF828ZPN.chr14.proc.bam" "ENCFF045OAB.chr14.proc.bam"
Geneid Chr Start End Strand Length ENCFF363HBZ.chr14.proc.bam ENCFF398QLV.chr14.proc.bam ENCFF828ZPN.chr14.proc.bam ENCFF045OAB.chr14.proc.bam
nk_merged_macs3_1 chr14 19161216 19161474 . 259 12 8 36 16
nk_merged_macs3_2 chr14 19161804 19162012 . 209 9 6 45 32
nk_merged_macs3_3 chr14 19239901 19240289 . 389 22 18 64 38
nk_merged_macs3_4 chr14 19384255 19384509 . 255 3 11 35 27
nk_merged_macs3_5 chr14 19488513 19488925 . 413 26 17 95 71
nk_merged_macs3_6 chr14 20305439 20306101 . 663 339 372 143 97
nk_merged_macs3_7 chr14 20332839 20333570 . 732 262 228 199 135
nk_merged_macs3_8 chr14 20342750 20343788 . 1039 2555 2424 1774 1226
We should remove the first line starting with #, as it can interfere with the way R reads in data:
awk '(NR>1)' nk_merged_peaks_macs3.counts > nk_merged_peaks_macs3.counts.tsv
Differential Accessibility
Please note that in the following exercise we use a counts table generated using a different peak set, hence some small differences to peaks called during the course may be present.
You can continue working in the atacseq/analysis/counts directory. This directory contains merged peaks called earlier using macs3 callpeak as well as count tables derived from summarising of non-subset data (we won’t need the count tables for this exercise). We will use file nk_merged_peaks_macs3.counts and annotation libraries, which are preinstalled. We access them via a module R_packages.
We now load R and packages:
module load R_packages/4.1.1
We activate R console upon typing R in the terminal.
We begin by loading necessary libraries:
library(edgeR)
library(EDASeq)
library(GenomicAlignments)
library(GenomicFeatures)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
library(wesanderson)
library(Hmisc)
library(dplyr)
txdb = TxDb.Hsapiens.UCSC.hg38.knownGene
ff = FaFile("/proj/epi2023/atacseq_proc/hg38ucsc/hg38.fa")
We can read in the data, format it and define experimental groups:
cnt_table = read.table("nk_merged_peaks_macs3.counts", sep="\t", header=TRUE, blank.lines.skip=TRUE)
rownames(cnt_table)=cnt_table$Geneid
#update colnames of this count table
colnames(cnt_table)=c("Geneid","Chr","Start","End","Strand","Length","ENCFF363HBZ","ENCFF398QLV","ENCFF045OAB","ENCFF828ZPN")
groups = factor(c(rep("NK",2),rep("NKstim",2)))
#this data frame contains only read counts to peaks on assembled chromosomes
reads.peak = cnt_table[,c(7:10)]
We now prepare data with GC content of the peak regions for GC-aware normalisation.
gr = GRanges(seqnames=cnt_table$Chr, ranges=IRanges(cnt_table$Start, cnt_table$End), strand="*", mcols=data.frame(peakID=cnt_table$Geneid))
peakSeqs = getSeq(x=ff, gr)
gcContentPeaks = letterFrequency(peakSeqs, "GC",as.prob=TRUE)[,1]
#divide into 20 bins by GC content
gcGroups = Hmisc::cut2(gcContentPeaks, g=20)
mcols(gr)$gc = gcContentPeaks
Figure below shows that the accessibility measure of a particular genomic region is associated with its GC content. However, the slope and shape of the curves may differ between samples, which indicates that GC content effects are sample–specific and can therefore bias between–sample comparisons.
To visualise GC bias in peaks:
lowListGC = list()
for(kk in 1:ncol(reads.peak)){
set.seed(kk)
lowListGC[[kk]] = lowess(x=gcContentPeaks, y=log1p(reads.peak[,kk]), f=1/10)
}
names(lowListGC)=colnames(reads.peak)
dfList = list()
for(ss in 1:length(lowListGC)){
oox = order(lowListGC[[ss]]$x)
dfList[[ss]] = data.frame(x=lowListGC[[ss]]$x[oox], y=lowListGC[[ss]]$y[oox], sample=names(lowListGC)[[ss]])
}
dfAll = do.call(rbind, dfList)
dfAll$sample = factor(dfAll$sample)
p1.1 = ggplot(dfAll, aes(x=x, y=y, group=sample, color=sample)) +
geom_line(size = 1) +
xlab("GC-content") +
ylab("log(count + 1)") +
theme_classic()
pdf("GCcontent_peaks.pdf")
## plot just the average GC content
p1.1
dev.off()
Counts vs GC contents in ATAC-seq peaks.
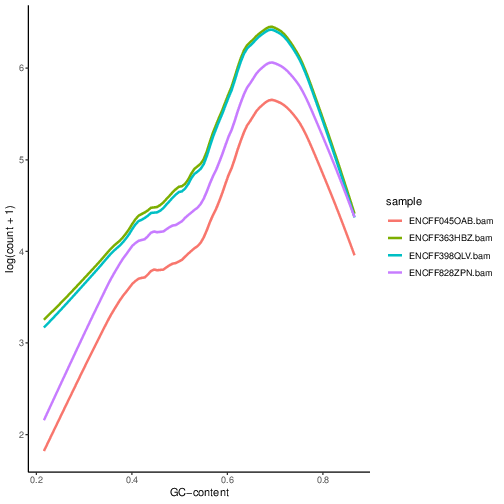
We can see that GC content has an effect on counts within the peaks.
We have seen from analyses presented on lecture slides and in https://www.biorxiv.org/content/10.1101/2021.01.26.428252v2
that full quantile normalisation (FQ-FQ) implemented in package EDASeq is one of the methods which can mitigate the GC bias in detection of DA regions.
We’ll detect differentially accessible regions using edgeR. We will input the normalised GC content as an offset to edgeR.
To calculate the offsets, which correct for library size as well as GC content (full quantile normalisation in both cases):
reads.peak=as.matrix(reads.peak)
dataOffset = withinLaneNormalization(reads.peak,y=gcContentPeaks,num.bins=20,which="full",offset=TRUE)
dataOffset = betweenLaneNormalization(reads.peak,which="full",offset=TRUE)
We now use the statistical framework of edgeR. We do not perform the internal normalisation (TMM) as usually, and instead we provide the offsets calculated by EDASeq.
design = model.matrix(~groups)
d = DGEList(counts=reads.peak, group=groups)
keep = filterByExpr(d)
> summary(keep)
Mode FALSE TRUE
logical 21 54743
d=d[keep,,keep.lib.sizes=FALSE]
d$offset = -dataOffset[keep,]
d.eda = estimateGLMCommonDisp(d, design = design)
d.eda = estimateGLMCommonDisp(d, design = design)
fit = glmFit(d.eda, design = design)
lrt.EDASeq = glmLRT(fit, coef = 2)
DA_res=as.data.frame(topTags(lrt.EDASeq, nrow(lrt.EDASeq$table)))
The top DA peaks in stimulated vs non-stimulated NK cells:
> head(DA_res)
logFC logCPM LR PValue FDR
nk_merged_macs3_30535 6.404743 5.289554 442.7384 2.744592e-98 2.274389e-93
nk_merged_macs3_14734 6.403253 4.939915 432.3314 5.052034e-96 2.093260e-91
nk_merged_macs3_16907 6.199114 4.881266 415.2994 2.573898e-92 7.109792e-88
nk_merged_macs3_43844 7.262906 4.361125 398.6341 1.092157e-88 2.262621e-84
nk_merged_macs3_18163 5.626103 6.097144 397.4212 2.005906e-88 3.324509e-84
nk_merged_macs3_46357 -5.894601 5.021572 392.9164 1.918571e-87 2.649803e-83
Let’s add more peak information:
DA_res$Geneid = rownames(DA_res)
DA.res.coords = left_join(DA_res,cnt_table[1:4],by="Geneid")
> head(DA.res.coords)
logFC logCPM LR PValue FDR Geneid
1 6.404743 5.289554 442.7384 2.744592e-98 2.274389e-93 nk_merged_macs3_30535
2 6.403253 4.939915 432.3314 5.052034e-96 2.093260e-91 nk_merged_macs3_14734
3 6.199114 4.881266 415.2994 2.573898e-92 7.109792e-88 nk_merged_macs3_16907
4 7.262906 4.361125 398.6341 1.092157e-88 2.262621e-84 nk_merged_macs3_43844
5 5.626103 6.097144 397.4212 2.005906e-88 3.324509e-84 nk_merged_macs3_18163
6 -5.894601 5.021572 392.9164 1.918571e-87 2.649803e-83 nk_merged_macs3_46357
Chr Start End
1 chr17 642297 643906
2 chr11 86292675 86294054
3 chr12 24838503 24839731
4 chr2 157477051 157477910
5 chr12 68155671 68157629
6 chr2 241985117 241985981
We can now save the results:
write.table(DA.res.coords, "nk_DA_stim_vs_ctrl.tsv", quote = FALSE, sep = "\t",
eol = "\n", na = "NA", dec = ".", row.names = FALSE,
col.names = TRUE, fileEncoding = "")
We can check how well the GC correction worked:
gcGroups.sub=gcGroups[keep]
dfEdgeR = data.frame(logFC=log(2^lrt.EDASeq$table$logFC), gc=gcGroups.sub)
pedgeR = ggplot(dfEdgeR) +
aes(x=gc, y=logFC, color=gc) +
geom_violin() +
geom_boxplot(width=0.1) +
scale_color_manual(values=wesanderson::wes_palette("Zissou1", nlevels(gcGroups), "continuous")) +
geom_abline(intercept = 0, slope = 0, col="black", lty=2) +
ylim(c(-1,1)) +
ggtitle("log2FCs in bins by GC content, FQ-FQ normalisation") +
xlab("GC-content bin") +
theme_bw()+
theme(aspect.ratio = 1)+
theme(axis.text.x = element_text(angle = 45, vjust = .5),
legend.position = "none",
axis.title = element_text(size=16))
ggsave(filename="log2FC_vs_GCcontent.EDAseq.pdf",plot=pedgeR ,path=".",device="pdf")
Dependence of log2FC on GC content in ATAC-seq.
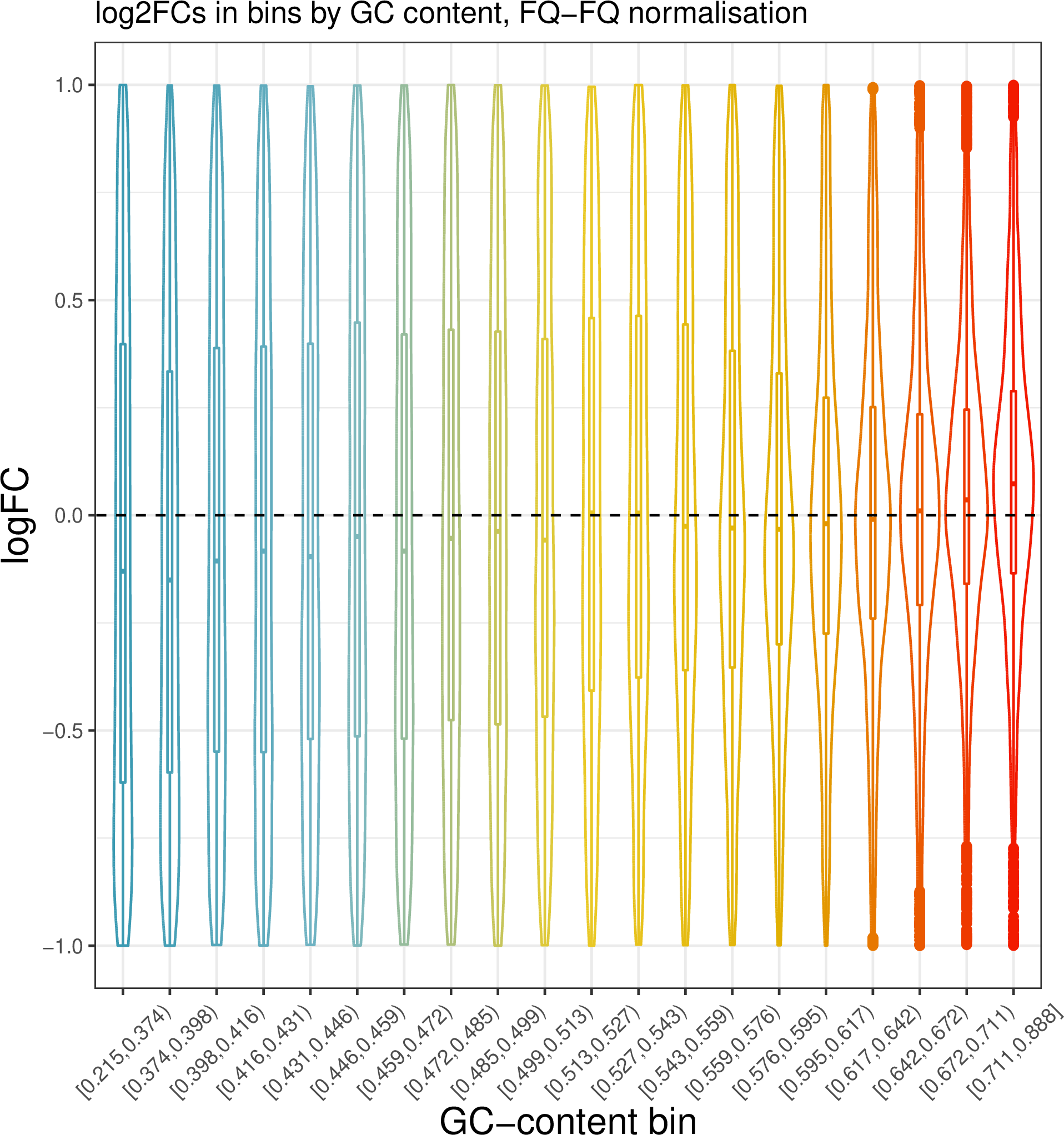
It seems that FQ-FQ normalisation did not completely remove the effect of GC content on log2FC in thie dataset. However, these effects are somewhat mitigated, you can compare this plot to one obtained by using the standard TMM normalisation.
Dependence of log2FC on GC content in ATAC-seq in non-GC corrected data.
d = DGEList(counts=reads.peak, group=groups)
keep = filterByExpr(d)
d=d[keep,,keep.lib.sizes=FALSE]
d = calcNormFactors(d)
#subset GRanges object for logFC binning
gr.sub=gr[keep,]
gcGroups.sub=gcGroups[keep]
d = estimateDisp(d, design)
fit <- glmFit(d, design)
lrt.tmm <- glmLRT(fit)
dfEdgeR = data.frame(logFC=log(2^lrt.tmm$table$logFC), gc=gcGroups.sub)
pedgeR <- ggplot(dfEdgeR) +
aes(x=gc, y=logFC, color=gc) +
geom_violin() +
geom_boxplot(width=0.1) +
theme_bw()+ theme(aspect.ratio = 1)+
scale_color_manual(values=wesanderson::wes_palette("Zissou1", nlevels(gcGroups), "continuous")) +
geom_abline(intercept = 0, slope = 0, col="black", lty=2) +
ylim(c(-1,1)) +
ggtitle("log2FCs in bins by GC content, TMM normalisation") +
theme(axis.text.x = element_text(angle = 45, vjust = .5),
legend.position = "none",
axis.title = element_text(size=16))
ggsave(filename="log2FC_vs_GCcontent.TMM.pdf",plot=pedgeR ,path=".",device="pdf")
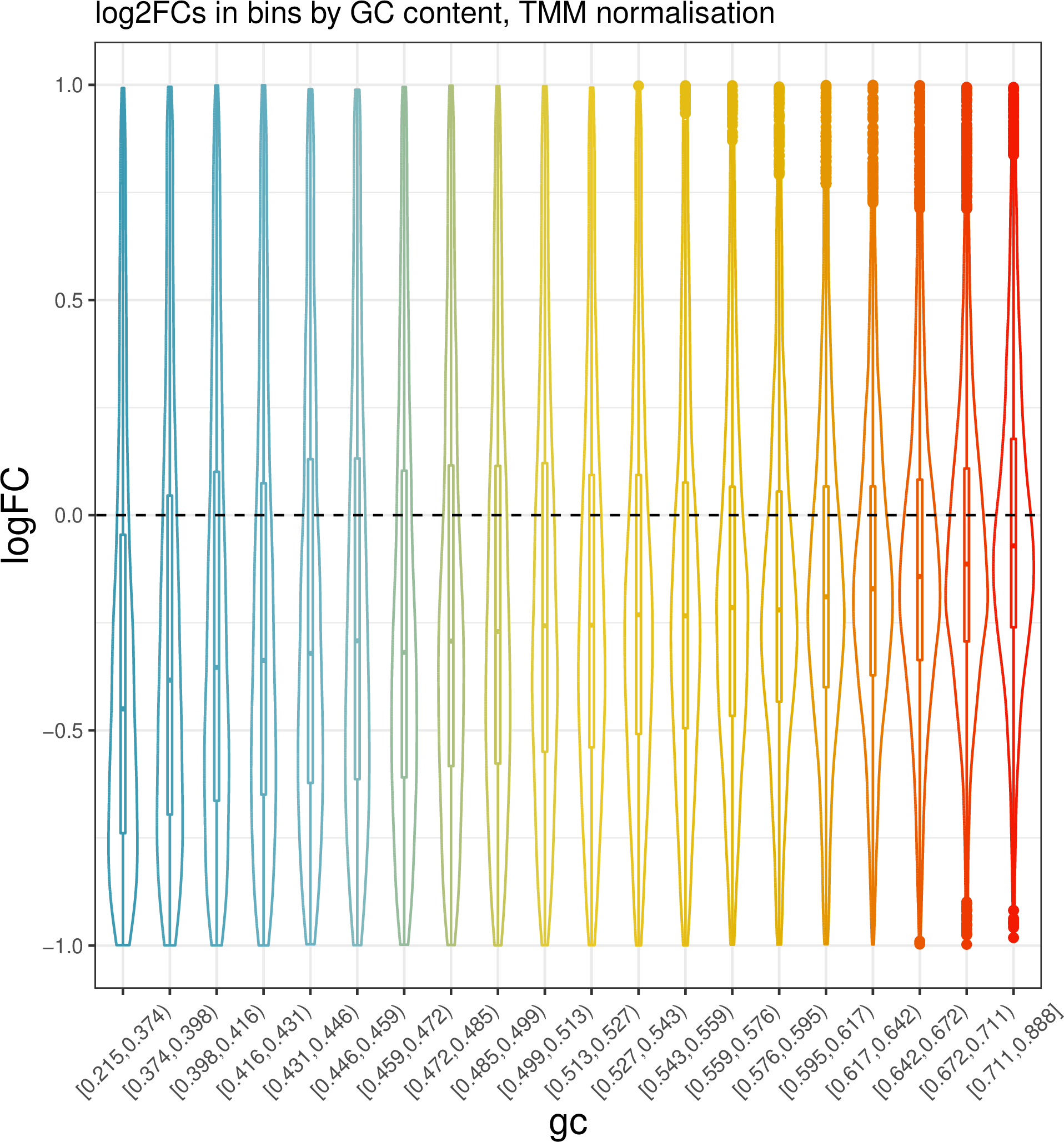
Part of the reason why the GC effects are not completely removed in this case may be that the fold change calculation/ DA analysis is not performed on properly replicated data; we should have at least 3 replicates per condition, and we only have two.