Transcription Factor Footprinting
Learning outcomes
detect transcription factor binding signatures in ATAC-seq data
|
Introduction
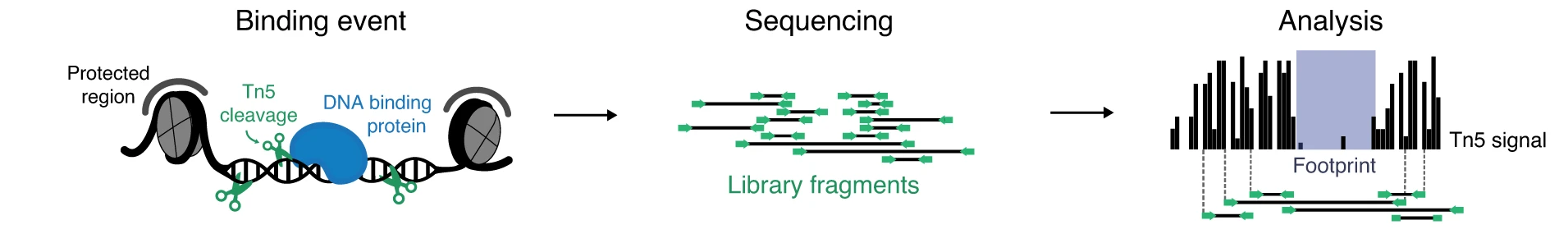
Transcription factor (TF) footprinting allows for the prediction of binding of a TF at a particular locus. This is because the DNA bases that are directly bound by the TF are actually protected from transposition while the DNA bases immediately adjacent to TF binding are accessible (Figure 1.). This results in footprints: defined regions of decreased signal strength within larger regions of high signal.
While ATAC-seq can uncover accessible regions where transcription factors (TFs) might bind, reliable identification of specific TF binding sites (TFBS) still relies on chromatin immunoprecipitation methods such as ChIP-seq. ChIP-seq methods require high input cell numbers, are limited to one TF per assay, and are further restricted to TFs for which antibodies are available.
Another caveat of TF footprinting is that not all TFs leave footprints detectable in ATAC-seq data, and not all footprints for given TF can be detected.
In this tutorial we use TOBIAS to detect TF binding signatures in ATAC-seq data (Bentsen et al 2020).
Data
We will use data that come from publication Batf-mediated epigenetic control of effector CD8+ T cell differentiation (Tsao et al 2022). These are ATAC-seq libraries (in duplicates) prepared to analyse chromatin accessibility status in murine CD8+ T lymphocytes prior to and upon Batf knockout.
The response of naive CD8+ T cells to their cognate antigen involves rapid and broad changes to gene expression that are coupled with extensive chromatin remodeling. Basic leucine zipper ATF-like transcription factor Batf is essential for the early phases of the process.
We will use data from in vivo experiment.
SRA sample accession numbers are listed in Table 1.
No |
Accession |
Sample Name |
Description |
|---|---|---|---|
1 |
SRR17296554 |
B1_WT_Batf-floxed_Cre_P14 |
WT Batf |
2 |
SRR17296555 |
B2_WT_Batf-floxed_Cre_P14 |
WT Batf |
3 |
SRR17296556 |
A1_Batf_cKO_P14 |
KO Batf |
4 |
SRR17296557 |
A2_Batf_cKO_P14 |
KO Batf |
We will use precomputed tracks and perform the last step: footprints detection.
We will use motifs of selected TF, to shorten the computation time.
We will inspect the results of footprinting of a comprehensive TF landscape of non-redundant vertebrate TF motif collection from JASPAR .
The starting point for this analyses are bam files with alignments merged across replicates i.e. one bam file per condition. This is done to increase read depth.
Setting-up
Starting at atacseq/analysis we will create a dedicated directory and copy necessary files.
mkdir TF_footprinting
cd TF_footprinting
cp /sw/courses/epigenomics/2025/lab-prep/cp_TFftprnt.sh .
bash cp_TFftprnt.sh
Detection of TF Binding Signatures
TOBIAS workflow consists of three stages:
Correction for the Tn5 transposase insertion sequence bias using
ATACorrect;Identify regions of protein binding in open chromatin (within peaks) using
ScoreBigwig;Calculate TF binding by combining footprint scores and TF binding motif information using
BINDetect.
ATACorrect and ScoreBigwig
We have precomputed these tracks, as it takes time and CPU resources.
TOBIAS ATACorrect --bam B_WT_merged_replicates.sorted.bam --genome GRCm39/Mus_musculus.GRCm39.dna.primary_assembly.fa --peaks genrich_joint_peaks_merged.Allpeaks_annot.Ensembl.bed --outdir TF_footprinting/tracks/B_WT/Footprint/
TOBIAS ScoreBigwig --signal Footprint/B_WT_merged_replicates.sorted_corrected.bw --regions genrich_joint_peaks_merged.Allpeaks_annot.Ensembl.bed --output B_WT_footprints.bw
TF Binding Detection
We will run TOBIAS BINDetect in comparative mode where we compare TF footprints in BATF KO vs WT.
We need to set some paths first:
container_pth="/sw/courses/epigenomics/2025/software/singularity/agatasm-tobias-uropa.img"
file_fa="/sw/courses/epigenomics/2025/reference/GRCm39/Mus_musculus.GRCm39.dna.primary_assembly.fa"
motifs_file="JASPAR2024_selTFs_pfms_meme.txt"
peaks="genrich_joint_peaks_merged.Allpeaks_annot.Ensembl.bed"
footprnt_1="/sw/courses/epigenomics/2025/atacseq/tsao2022/proc_6ix2025/TF_footprinting/tracks/A_Batf_KO/Footprint/A_Batf_KO_footprints.bw"
footprnt_2="/sw/courses/epigenomics/2025/atacseq/tsao2022/proc_6ix2025/TF_footprinting/tracks/B_WT/Footprint/B_WT_footprints.bw"
smpl="Batf_KO_vs_WT_selTFs"
We will execute TOBIAS in a software container, so the command looks a bit different.
apptainer exec ${container_pth} TOBIAS BINDetect --motifs ${motifs_file} --signals ${footprnt_1} ${footprnt_2} --genome ${file_fa} --peaks ${peaks} --cores 6 --outdir ${smpl}/BINDdetect
Output description can be found at BINDetect documentation
You can view summary of the results obtained for all TFs in directory all_TFs. In particular, file bindetect_A_Batf_KO_footprints_B_WT_footprints.html contains an interactive volcano plot highlighting the TFs with most extreme differences in their footprint scores uopn Batf knock-out.

|
Batf motif is part of the JUN / FOS points cluster at the top left arm of the volcano. The results for one of its motifs are:
output_prefix name motif_id cluster total_tfbs A_Batf_KO_footprints_mean_score A_Batf_KO_footprints_bound B_WT_footprints_mean_score B_WT_footprints_bound A_Batf_KO_footprints_B_WT_footprints_change A_Batf_KO_footprints_B_WT_footprints_pvalue A_Batf_KO_footprints_B_WT_footprints_highlighted
BATF_MA1634.2 BATF MA1634.2 C_FOS::JUND 17254 0.33875 2304 0.38674 2747 -0.39584 1.07531E-169 True
References
Tsao, Hsiao-Wei, James Kaminski, Makoto Kurachi, R. Anthony Barnitz, Michael A. DiIorio, Martin W. LaFleur, Wataru Ise, et al. 2022. “Batf-Mediated Epigenetic Control of Effector CD8 + t Cell Differentiation.” Science Immunology 7 (68). https://doi.org/10.1126/sciimmunol.abi4919.
Bentsen, Mette, Goymann Philipp, Schultheis Hendrik, Klee Kathrin, Petrova Anastasiia, Wiegandt René, Fust Annika, Preussner Jens, Kuenne Carsten, Braun Thomas, Kim Johnny, Looso Mario 2020. “ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation” Nature Communications Vol. 11, No. 1 https://doi.org/10.1038/s41467-020-18035-1