ATAC-seq
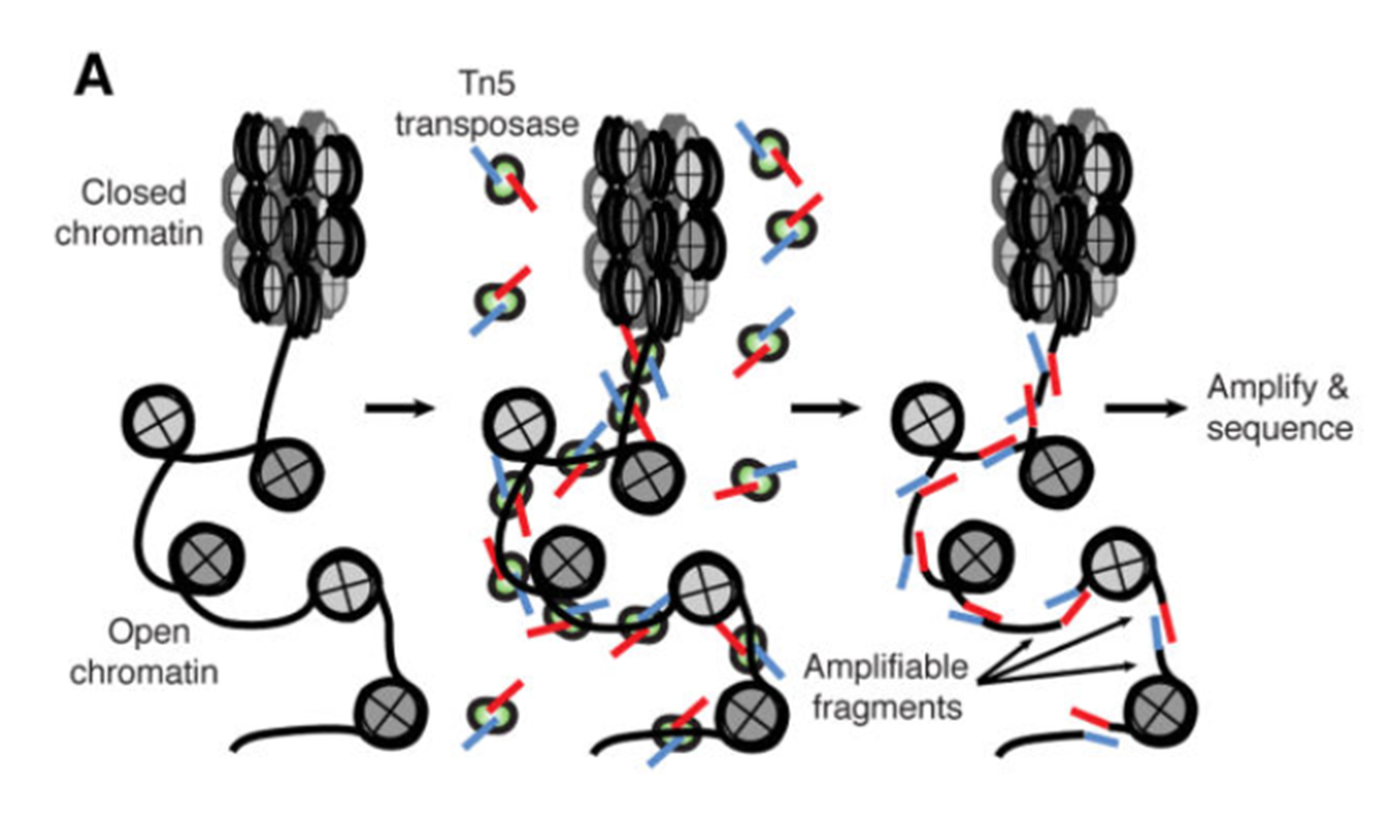
ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) is a method for determining chromatin accessibility across the genome. It utilizes a hyperactive Tn5 transposase to insert sequencing adapters into open chromatin regions. High-throughput sequencing then yields reads that indicate these regions of increased accessibility.
|
This tutorial is a continuation of ATAC-seq QC.
Learning outcomes
identify peaks in ATAC-seq data using different methods
calculate fraction of reads in peaks
generate counts table
This tutorial is a continuation of Data preprocessing, General QC, and ATACseq specifc QC.
Important
We assume that the environment and directory structure has been already set in Data preprocessing.
Data
We will use data that come from publication Batf-mediated epigenetic control of effector CD8+ T cell differentiation (Tsao et al 2022). These are ATAC-seq libraries (in duplicates) prepared to analyse chromatin accessibility status in murine CD8+ T lymphocytes prior to and upon Batf knockout.
The response of naive CD8+ T cells to their cognate antigen involves rapid and broad changes to gene expression that are coupled with extensive chromatin remodeling. Basic leucine zipper ATF-like transcription factor Batf is essential for the early phases of the process.
We will use data from in vivo experiment.
SRA sample accession numbers are listed in Table 1.
No |
Accession |
Sample Name |
Description |
|---|---|---|---|
1 |
SRR17296554 |
B1_WT_Batf-floxed_Cre_P14 |
WT Batf |
2 |
SRR17296555 |
B2_WT_Batf-floxed_Cre_P14 |
WT Batf |
3 |
SRR17296556 |
A1_Batf_cKO_P14 |
KO Batf |
4 |
SRR17296557 |
A2_Batf_cKO_P14 |
KO Batf |
We have processed the data, starting from raw reads. The reads were aligned to GRCm39 reference assembly using bowtie2 and subset to include alignments to chromosome 1 and 1% of reads mapped to chromosomes 2 to 5 and MT.
This allows you to see a realistic coverage of one selected chromosome and collect QC metrics while allowing shorter computing times.
In this workshop, we have previously filtered and quality-controlled the data (parts Data preprocessing, General QC, and ATACseq specifc QC).
Peak Calling
To find regions corresponding to potential open chromatin, we want to identify ATAC-seq “peaks” where reads have piled up to a greater extent than the background read coverage.
The tools which are currently used in major processing pipelines are Genrich and MACS3.
Genrich has a mode dedicated to ATAC-Seq (in which it creates intervals centered on transposase cut sites); it can leverage biological replicates in calculating significance; however, Generich is still not published;
MACS3 has more ATAC-seq oriented features than its predecessor MACS2, however, its main algorithm for peak detection is oriented towards peak calling in ChIP-seq experiments and does not take into account unique features of ATAC-seq data.
The differences between Genrich and MACS2/2 in the context of ATAC-seq data are discussed here.
In this tutorial we will use Genrich and MACS3. We will compare the results of peaks detected by Genrich and by MACS3, as used in major data processing pipelines (nf-core, ENCODE).
Shifting Alignments
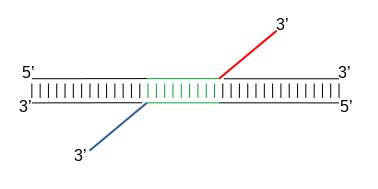
We have already discussed (and performed) this step in the ATACseq specifc QC tutorial. Briefly, the alignments are shifted to account for the duplication created as a result of DNA repair after Tn5-introduced DNA nicks.
When Tn5 cuts an accessible chromatin locus it inserts adapters separated by 9bp, see Figure 2. This means that to have the read start site reflect the centre of where Tn5 bound, the reads on the positive strand should be shifted 4 bp to the right and reads on the negative strand should be shifted 5 bp to the left as in Buenrostro et al. 2013.
|
To shift or not to shift? It, as always, depends on the downstream application.
If we use the ATAC-seq peaks for differential accessibility, and especially if we detect the peaks in the MACS3 “broad” mode, then shifting does not play any role: the “peaks” are hundred(s) of bps long, reads are summarised to these peaks / domains allowing a partial overlap, so 9 basepairs of difference in read position has a neglibile effect.
However, when we plan to use the data for any nucleosome-centric analysis (positioning at TSS or TF footprinting), shifting the reads allows to center the signal in peaks flanking the nucleosomes and not directly on the nucleosome. Typically, applications used for these analyses perform the read shifting, so we do not need to preprocess the input bam files.
More on peak calling and which parameters to choose. Historically, ATAC-seq data have been analysed by software developed for ChIP-seq, even though the two assays have different signal structure. The peak caller most widely used was MACS2 (as evidenced by number of citations), with a wide range of parameters. The discussion on why not all these parameter choices are optimal can be found in Gaspar 2018. In this exercise you will see MACS3 callpeak algorithm used in two different settings.
Genrich
We start this tutorial in directory atacseq/analysis/:
mkdir peaks
cd peaks
We need to link necessary files first.
mkdir genrich
cd genrich
# we link the pre-processed bam file
ln -s ../../../data_proc/SRR17296554.filt.chr1.bam
ln -s ../../../data_proc/SRR17296554.filt.chr1.bam.bai
# because we want to use the other replicate for peak calling, we link its files as well
ln -s ../../../data_proc/SRR17296555.filt.chr1.bam
ln -s ../../../data_proc/SRR17296555.filt.chr1.bam.bai
In these files, we removed all reference sequences other than chr1 from the bam header, as this is where our data is subset to. Genrich uses the reference sequence length from bam header in claculating the statistical significance, so retaining the original bam header would impair peak calling statistics.
Genrich requires bam files to be name-sorted rather than the default coordinate-sorted.
# in case not already loaded
module load bioinfo-tools
module load samtools/1.19
# sort the bam file by read name (required by Genrich)
samtools sort -n -o SRR17296554.filt.chr1.nsort.bam SRR17296554.filt.chr1.bam
samtools sort -n -o SRR17296555.filt.chr1.nsort.bam SRR17296555.filt.chr1.bam
Genrich can apply the read shifts when ATAC-seq mode -j is selected. We detect peaks by:
/sw/courses/epigenomics/ATACseq_bulk/software/Genrich/Genrich -j -t SRR17296554.filt.chr1.nsort.bam -o SRR17296554.chr1.genrich.narrowPeak
The output file produced by Genrich is in ENCODE narrowPeak format, listing the genomic coordinates of each peak called and various statistics.
chr start end name score strand signalValue pValue qValue peak
signalValue - Measurement of overall (usually, average) enrichment for the region.
pValue - Measurement of statistical significance (-log10). Use -1 if no pValue is assigned.
qValue - Measurement of statistical significance using false discovery rate (-log10). Use -1 if no qValue is assigned.
How many peaks were detected?
wc -l SRR17296554.chr1.genrich.narrowPeak
2860 SRR17296554.chr1.genrich.narrowPeak
SRR17296554.chr1.genrich.narrowPeak
You can inspect file contents:
head SRR17296554.chr1.genrich.narrowPeak
1 4566396 4566996 peak_0 874 . 524.450012 4.083697 -1 295
1 4818047 4818755 peak_1 739 . 523.147156 3.863627 -1 476
1 4855285 4856526 peak_2 1000 . 2765.520264 7.476175 -1 601
1 4877542 4879096 peak_3 1000 . 2765.986816 6.540341 -1 573
1 4927128 4929149 peak_4 1000 . 4235.729492 7.914668 -1 868
1 5152781 5154166 peak_5 1000 . 2653.163330 7.412836 -1 556
1 6283374 6283786 peak_6 549 . 226.311874 3.085280 -1 279
1 6284256 6285987 peak_7 1000 . 4862.475098 7.119522 -1 913
1 6452823 6453287 peak_8 579 . 268.770874 3.621369 -1 280
1 6476302 6477424 peak_9 1000 . 1561.405884 6.319389 -1 506
We can now process the remaining replicate:
/sw/courses/epigenomics/ATACseq_bulk/software/Genrich/Genrich -j -t SRR17296555.filt.chr1.nsort.bam -o SRR17296555.chr1.genrich.narrowPeak
Genrich can also call peaks for multiple replicates collectively. First, it analyzes the replicates separately, with p-values calculated for each. At each genomic position, the multiple replicates’ p-values are then combined by Fisher’s method. The combined p-values are converted to q-values, and peaks are output.
For comparison, use the joint replicate peak calling mode:
/sw/courses/epigenomics/ATACseq_bulk/software/Genrich/Genrich -j -t SRR17296554.filt.chr1.nsort.bam,SRR17296554.filt.chr1.nsort.bam -o WT_Batf.chr1.genrich.narrowPeak
How many peaks were detected by Genrich?
wc -l *narrowPeak
2860 SRR17296554.chr1.genrich.narrowPeak
2791 SRR17296555.chr1.genrich.narrowPeak
4661 WT_Batf.chr1.genrich.narrowPeak
It turns out Genrich detected more peaks in the joint mode, including in locations not picked up in neither of the individual libraries. Some of these locations have been also detected by MACS3 callpeak, see figures below. This requires further investigation and comparisons, for now however we can conclude that adding more replicates improves the sensitivity of peak calling. Upon inspection of alignments in bam files and peak calls, many of the peaks called only in joint replicate analysis tend to have lower signal-to-noise ratio than peaks detected also in individual replicates.
MACS
As mentioned in the introduction, MACS has been used with a variety of parametr choices to detect peaks in ATAC-seq data.
You can encounter various combinations of read input modes (BAM, BAMPE, BED and BEDPE; BED / BAM designaed for SE data used also for PE data), and peak calling modes (default “narrow” and optional “broad”). Some protocols shift reads (setting options extsize and shift) as for centring reads on the binding site in ChIP-seq peak calling workflows.
MACS3 broead peak
Here we choose the simplest approach: detection of regions with signal higher than expected (background modelled by Poisson distribution) in fragment pileups.
First, we create a separate directory for results obtained using MACS3:
mkdir -p ../macs3
cd ../macs3
module load MACS/3.0.0b1
We are now ready to call the peaks using macs3 callpeak. We will use the same file as for Genrich, and the genome size of 195154279 (length of chr 1).
ln -s ../../../data_proc/SRR17296554.filt.chr1.bam
macs3 callpeak --keep-dup all --nomodel --broad --broad-cutoff 0.1 -g 195154279 -f BAMPE -t SRR17296554.filt.chr1.bam -n SRR17296554.macs3_broad.bampe
This command is used in nf-core ataseq pipelines.
How many peaks were detected?
6668 SRR17296554.macs3_broad.bampe_peaks.broadPeak
We can see that the peak numbers are in the same ballpark, as for Genrich. We expect that some calls will be different, but more or less these results seem to go in line with one anoter.
Figures below use the same colour scheme, tracks from top:
processed bam files with coverage
gene models (indigo)
Genrich (dark magenta), peaks called on joint replictaes
Genrich (green), peaks for replicate 1
Genrich (green), peaks for replicate 2
macs3 callpeak (blue), broad peak, BAMPE, replicate 1

|

|

|

|
Upon inspecting the tracks, we notice that while the peaks with high signal-to-noise ratio are identifed by both methods (MACS3 broad BAMPE and genrich), some peaks are detected by single method. In this case some of the peaks detected by MACS3 do not seem to correspond to true signal.
This highlights the need to always inspect the data and peak calls. In many cases spurious calls may be mitigated by filtering.
Fraction of Reads in Peaks
Fraction of Reads in Peaks (FRiP) is one of key QC metrics of ATAC-seq data. According to ENCODE ATACseq data standards acceptable FRiP is >0.2. This value of course depends on the peak calling protocol, and as we have seen in the previous section, the results may vary …a lot. However, it is worth to keep in mind that any samples which show different value for this (and other) metric may be outliers problematic in the analysis.
To calculate FRiP we need alignment file (bam) and peak file (narrowPeak, bed).
Assuming we are in peaks:
mkdir frip
cd frip
We will use a tool called featureCounts from package Subread. This tool accepts genomic intervals in formats gtf/gff and saf. Let’s convert narrow/ broadPeak to saf:
ln -s ../genrich/SRR17296554.chr1.genrich.narrowPeak
awk -F $'\t' 'BEGIN {OFS = FS}{ $2=$2+1; peakid="genrich_Peak_"++nr; print peakid,$1,$2,$3,"."}' SRR17296554.chr1.genrich.narrowPeak > SRR17296554.chr1.genrich.saf
SRR17296554.chr1.genrich.saf
genrich_Peak_1 1 4566397 4566996 .
genrich_Peak_2 1 4818048 4818755 .
genrich_Peak_3 1 4855286 4856526 .
genrich_Peak_4 1 4877543 4879096 .
genrich_Peak_5 1 4927129 4929149 .
genrich_Peak_6 1 5152782 5154166 .
genrich_Peak_7 1 6283375 6283786 .
genrich_Peak_8 1 6284257 6285987 .
genrich_Peak_9 1 6452824 6453287 .
genrich_Peak_10 1 6476303 6477424 .
We can now summarise reads:
ln -s ../genrich/SRR17296554.filt.chr1.bam
module load subread/2.0.3
featureCounts -p -F SAF -a SRR17296554.chr1.genrich.saf --fracOverlap 0.2 -o SRR17296554.peaks_genrich.counts SRR17296554.filt.chr1.bam
This command has produced reads summarised within each peak (which we won’t use at this stage) and a summary file SRR17296554.peaks_genrich.counts.summary which contains values we are interested in:
Status SRR17296554.filt.chr1.bam
Assigned 3659169
Unassigned_Unmapped 0
Unassigned_Read_Type 0
Unassigned_Singleton 0
Unassigned_MappingQuality 0
Unassigned_Chimera 0
Unassigned_FragmentLength 0
Unassigned_Duplicate 0
Unassigned_MultiMapping 0
Unassigned_Secondary 0
Unassigned_NonSplit 0
Unassigned_NoFeatures 5473213
Unassigned_Overlapping_Length 6387
Unassigned_Ambiguity 0
3659169/(3659169+5473213+6387) = 0.4004 (i.e. 40.0%)
featureCounts reported in the output to the screen (STDOUT) that 40.0% reads fall within peaks, and this is FRiP for sample SRR17296554.
Note
Please note that the values of FRiP depend on the peak calling protocol. Methods producing many more peaks (such as used in the ENCODE pipeline) will result in higher FRiP values. Thus, when comparing values in given experiment to data standards (such as from ENCODE), it is important to know the details of data processing used to derive given statistic.
Count Table
In preparation for the Differential Accessibility analysis we need to create counts table: reads mapped to merged peaks detected in all samples in the data set, to have a consensus peak set we can compare between conditions.
To get the merged peaks, we can simply merge the peaks detected individually in each sample, or use some more complex scheme (e.g. only merge peaks reproducible between replicates etc.)
In this example we will merge peaks detected by genrich in joint mode. We will work with peaks on chr1, to shorten computation time.
Starting at peaks:
mkdir counts
cd counts
module load BEDOPS/2.4.41
bedops -m ../genrich/WT_Batf.chr1.genrich.narrowPeak /proj/epi2025/atacseq_proc/A_Batf_cKO_P14.genrich_joint.chr1.narrowPeak > AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.bed
How many peaks?:
4904 AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.bed
The format of AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.bed is a very simple, 3-field BED file. Let’s add peaks ids and convert it to saf (id-chr-start-end-strand):
awk -F $'\t' 'BEGIN {OFS = FS}{ $2=$2+1; peakid="genrich_merged_"++nr; print peakid,$1,$2,$3,"."}' AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.bed > AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.saf
This file can now be used to generate counts table, as above. This command counts each read seprately (rather than as a pair / fragment) because each Tn5 transposition is an independent event.
ln -s ../genrich/SRR17296554.filt.chr1.bam
ln -s ../genrich/SRR17296555.filt.chr1.bam
ln -s ../../../data_proc/SRR17296556.filt.chr1.bam
ln -s ../../../data_proc/SRR17296557.filt.chr1.bam
featureCounts -p -F SAF -a AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.saf --fracOverlap 0.2 -o AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.counts SRR17296554.filt.chr1.bam SRR17296555.filt.chr1.bam SRR17296556.filt.chr1.bam SRR17296557.filt.chr1.bam
Let’s take a look inside the counts table using head AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.counts.
nk_merged_peaks_macs3.counts
head AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.counts
# Program:featureCounts v2.0.3; Command:"featureCounts" "-p" "-F" "SAF" "-a" "AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.saf" "--fracOverlap" "0.2" "-o" "AB_Batf_KO_invivo.genrich_joint.merged_peaks.chr1.counts" "SRR17296554.filt.chr1.bam" "SRR17296555.filt.chr1.bam" "SRR17296556.filt.chr1.bam" "SRR17296557.filt.chr1.bam"
Geneid Chr Start End Strand Length SRR17296554.filt.chr1.bam SRR17296555.filt.chr1.bam SRR17296556.filt.chr1.bam SRR17296557.filt.chr1.bam
genrich_merged_1 1 3051361 3052202 . 842 201 152 192 213
genrich_merged_2 1 4566251 4567370 . 1120 405 355 381 395
genrich_merged_3 1 4817899 4818868 . 970 394 261 419 368
genrich_merged_4 1 4838532 4839221 . 690 181 124 137 225
genrich_merged_5 1 4840062 4840755 . 694 209 131 236 187
genrich_merged_6 1 4845165 4845849 . 685 92 72 147 168
genrich_merged_7 1 4850233 4850769 . 537 167 161 268 214
genrich_merged_8 1 4855217 4856703 . 1487 1822 1508 2001 2089
This file can be read in as a table in R.
We can now follow with downstream analyses: Peak Annotation, Differential Accessibility
References
Tsao, Hsiao-Wei, James Kaminski, Makoto Kurachi, R. Anthony Barnitz, Michael A. DiIorio, Martin W. LaFleur, Wataru Ise, et al. 2022. “Batf-Mediated Epigenetic Control of Effector CD8 + t Cell Differentiation.” Science Immunology 7 (68). https://doi.org/10.1126/sciimmunol.abi4919.