Detection of broad peaks from ChIP-seq data
Please note this tutorial is more than 4 years old. It uses older tools versions and has not been recently tested.
Learning outcomes
be able to assess the quality of ChIP-seq data for factors with broad occupancy pattern
be able to detect regions of enrichment for factors with broad occupancy pattern
Requirements
MACS 3.0.0a6epic2 0.0.52R version 4.0.4 (2021-02-15) -- "Lost Library Book"csaw 1.24.3and its dependencies
Bioconductor packages required for annotation:
org.Hs.eg.dbTxDb.Hsapiens.UCSC.hg38.knownGene
For details please consult Dependencies.
Please note that this lab consists of three parts:
calling broad peaks using
MACS(Uppmax);calling broad peaks using
epic2(Uppmax); andfinding enriched genomic windows using
csawinR(Uppmax / local).
Notes on software
We provide a conda environment to run epic2. This package proved a bit tricky to install because of dependency incompatibilities. To find how this environment was constructed, please visit Dependencies.
Please note that this workflow has been tested using R 4.0.4 and csaw 1.24.3 on Uppmax only.
Data
We will use ChIP-seq of H3K79me2 from ENCODE. H3K79me2 is enriched at active promoters and linked to transcriptional activation as it tends to accumulate in transcribed regions of active genes.
The samples are from GM23338 cell line (human induced pluripotent stem cell from skin fibroblasts) before and after stimulation with doxycycline to form bipolar neurons.
These data are paired-end with read length 76 bp (2x76) and consist of duplicates of H3K79me2 ChIP and matching input samples. Raw data was processed using ENCODE3 pipeline and mapped to GRCh38 genome assembly. We have further processed the data to remove unwanted signal and subset to chromosomes 1 and 2, as in the tutorial ChIPseq data processing. Accession details are summarised in Table 1.
Sample |
ENCODE Accession |
Replicate |
|---|---|---|
GM23338, H3K79me2 ChIP |
ENCFF263XBT |
rep 1 |
GM23338, H3K79me2 ChIP |
ENCFF531WSU |
rep 2 |
GM23338, input |
ENCFF992ZBS |
rep 1 |
GM23338, input |
ENCFF237PRF |
rep 2 |
neuro GM23338, H3K79me2 ChIP |
ENCFF395DAJ |
rep 1 |
neuro GM23338, H3K79me2 ChIP |
ENCFF806KRA |
rep 2 |
neuro GM23338, input |
ENCFF956GLJ |
rep 1 |
neuro GM23338, input |
ENCFF687LIL |
rep 2 |
We will detect occupancy regions from one sample only, and compare the results to other samples processed earlier.
Quality control
As always, one should start the analysis from assesment of data quality. This is already performed to save time, and the plots and metrics are included below.
Cross-correlation and related metrics
The files discussed in this section can be accessed at
/proj/epi2025/broad_peaks/results/qc
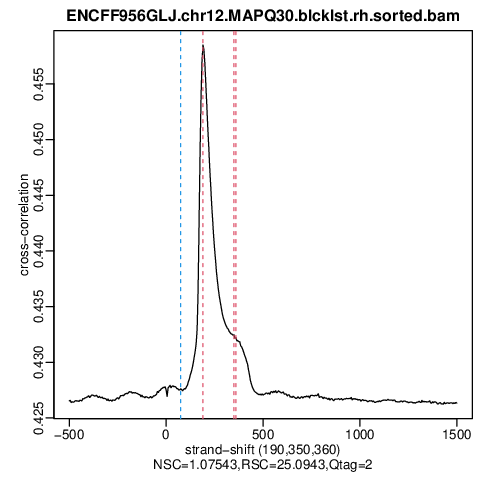
These metrics have been developed with application to point source (i.e. TFs and narrow histone modifications) ChIP-seq in mind, and you can see that the results for broad domains are not as easy to interpret as for point source factors. Below are cross correlation plots for the IP and input you are going to use for the exercise.
Already from these plots alone it is evident that the data has some quality issues. At this point you should be able to identify them.
K79me2 ChIP (ENCFF395DAJ) |
input (ENCFF956GLJ) |
|---|---|

|
|
The cross correlation profile of factors with broad occupancy patterns is not going to be as sharp as for TFs, and the values of NSC and RSC tend to be lower, which does not mean that the ChIP failed. In fact, the developers of the tool do not recommend using the same NSC / RSC values as quality cutoffs for broad marks. However, input samples should not display signs of enrichment, as is the case here.
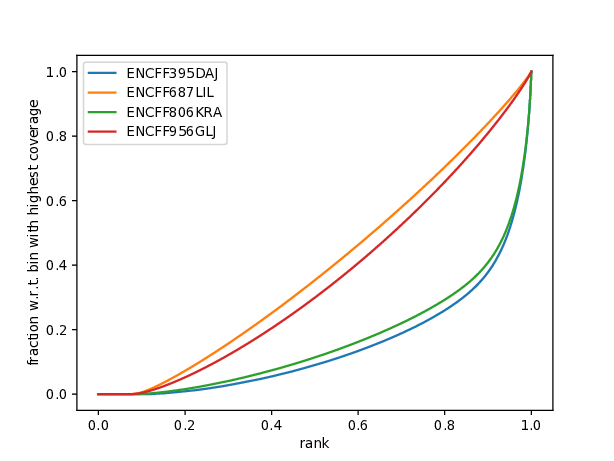
Cumulative enrichment
Another plot worth examining is cumulative enrichment (aka fingerprint from deepTools):
all samples |
|---|
|
You can see that even though the cross correlation metrics don’t look great, a significant enrichment can be observed for the ChIP samples (ENCFF395DAJ, ENCFF806KRA), and not for the input samples.
Broad peak calling using MACS
MACS: Model-based Analysis for ChIP-Seq is one of the leading peak calling algorithms. We will use MACS version 3, which is still under active development and hasn’t been officially released.
You will call peaks using sample GM23338 neuro - H3K79me2 ChIP ENCFF395DAJ and its matching input ENCFF956GLJ.
Effective genome size for chr 1 and 2 in hg38 is 4.9e8.
mkdir -p analysis/macs3
cd analysis/macs3
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338/ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338//ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam
module load bioinfo-tools #if needed
module load MACS/3.0.0a6
macs3 callpeak --broad \
-t ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam \
-c ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam \
-f BAMPE -g 04.9e8 --broad-cutoff 0.1 -n neuroGM23338_macs3_rep1
The main difference here, in comparison to detecting narrow peaks, is using the options --broad --broad-cutoff 0.1. With the option --broad on, MACS will try to composite broad regions in BED12 (gene-model-like format) by putting nearby highly enriched regions into a broad region with loose cutoff. The broad region is controlled by another cutoff through --broad-cutoff. If -p is set, this is a p-value cutoff, otherwise, it’s a q-value (FDR) cutoff.
Because we use PE data, there is no need to build a model to estimate fragment length (similar to cross correlation) necessary for extending the SE reads. We know precisely how long each fragment is because its both ends are sequenced and mapped to the reference. Estimating fragment length from cross-correlation of broad domain ChIP data is problematic and sometimes may result in incorrect values.
You can now inspect the results in the output folder macs3. The structure is alike the output for calling narrow peaks. The file *.broadPeak is in BED6+3 format which is similar to narrowPeak file used for point-source factors, except for missing the 10th column for annotating peak summits. Look at MACS repository homepage for details.
The meaning of columns in NAME_peaks.xls files:
- chr
chromosome name
- start
start position of peak
- end
end position of peak
- length
length of peak region
- pileup
pileup height at peak summit
- -log10(pvalue)
-log10(pvalue) for the peak summit (e.g. pvalue =1e-10, then this value should be 10)
- fold_enrichment
fold enrichment for this peak summit against random Poisson distribution with local lambda
- -log10(qvalue)
-log10(qvalue) at peak summit
- name
peak id
Let’s take a look at another format of the output broadPeak. It is a derivative of bed format and thus compatible with major genome browsers (IGV, UCSC Genome Browser) and easier to work with because it does not contain a long header.
This is an example:
head neuroGM23338_macs3_rep1_peaks.broadPeak
chr1 777491 778262 neuroGM23338_macs3_rep1_peak_1 34 . 3.542 4.93525 3.48401
chr1 779812 780867 neuroGM23338_macs3_rep1_peak_2 10 . 2.28884 2.27839 1.03252
chr1 782000 784521 neuroGM23338_macs3_rep1_peak_3 17 . 2.6654 3.01765 1.70342
chr1 820548 826643 neuroGM23338_macs3_rep1_peak_4 36 . 3.5486 5.10182 3.65624
chr1 828271 830128 neuroGM23338_macs3_rep1_peak_5 34 . 3.4958 4.87316 3.42798
chr1 831350 833671 neuroGM23338_macs3_rep1_peak_6 22 . 2.7518 3.55309 2.20097
chr1 882552 890194 neuroGM23338_macs3_rep1_peak_7 34 . 3.21783 4.86863 3.43262
chr1 925794 926897 neuroGM23338_macs3_rep1_peak_8 18 . 2.71963 3.12803 1.80546
chr1 957085 959246 neuroGM23338_macs3_rep1_peak_9 60 . 4.54986 7.61848 6.03443
chr1 999291 999914 neuroGM23338_macs3_rep1_peak_10 16 . 2.63811 2.95948 1.65064
The meaning of columns in NAME.broadPeak files:
- chrom
Name of the chromosome (or contig, scaffold, etc.).
- chromStart
The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0.
- chromEnd
The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromStart=0, chromEnd=100, and span the bases numbered 0-99. If all scores were “0” when the data were submitted to the DCC, the DCC assigned scores 1-1000 based on signal value. Ideally the average signalValue per base spread is between 100-1000.
- name
Name given to a region (preferably unique). Use “.” if no name is assigned.
- score
Indicates how dark the peak will be displayed in the browser (0-1000).
- strand
+/- to denote strand or orientation (whenever applicable). Use “.” if no orientation is assigned.
- signalValue
Measurement of overall (usually, average) enrichment for the region.
- pvalue
Measurement of statistical significance (-log10). Use -1 if no pValue is assigned.
- qValue
Measurement of statistical significance using false discovery rate (-log10). Use -1 if no qValue is assigned.
How many peaks were identified in replicate 1?
wc -l neuroGM23338_macs3_rep1_peaks.broadPeak
6826 neuroGM23338_macs3_rep1_peaks.broadPeak
Hint
You can also copy the results from
/proj/epi2025/broad_peaks/results/macs3/neuroGM23338
This is a preliminary peak list, and in case of broad domains, it often needs some processing or filtering.
Let’s select the detected domains reproducible in both replicates. First, let’s create a subdirectory peaks, and link the results of broad peak calling. Then we select the first 6 columns of broadPeak to create files in BED-6 format, which are ready for use by bedtools. After completing the tutorial on data processing you should be able to find the peaks reproducible between the replicates. How many of them can we identify?
mkdir peaks
cd peaks
ln -s /proj/epi2025/broad_peaks/results/macs3/neuroGM23338/neuroGM23338_macs3_rep1_peaks.broadPeak
ln -s /proj/epi2025/broad_peaks/results/macs3/neuroGM23338/neuroGM23338_macs3_rep2_peaks.broadPeak
#make bed
cut -f 1-6 neuroGM23338_macs3_rep1_peaks.broadPeak >neuroGM23338_macs3_rep1_peaks.bed
cut -f 1-6 neuroGM23338_macs3_rep2_peaks.broadPeak >neuroGM23338_macs3_rep2_peaks.bed
Select reproducible peaks (MACS).
#intersect bed files
module load bioinfo-tools #if needed
module load BEDTools/2.29.2
bedtools intersect -a neuroGM23338_macs3_rep1_peaks.bed -b neuroGM23338_macs3_rep2_peaks.bed -f 0.50 -r > peaks_macs3_neuroGM23338.chr12.bed
#how many peaks which overlap?
wc -l peaks_macs3_neuroGM23338.chr12.bed
2679 peaks_macs3_neuroGM23338.chr12.bed
Visual inspection of the peaks (MACS)
You will use IGV for this step, and it is recommended that you run it locally on your own computer. Please load hg38 reference genome.
Required files are:
ChIP
ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bamand matchingbaiinput
ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bamand matchingbaisignal domains
neuroGM23338_macs3_rep1_peaks.broadPeakreproducible signal domains
peaks_macs3_neuroGM23338.chr12.bed
Hint
You can access the bam and bai files from
/proj/epi2025/broad_peaks/data/neuron_GM23338
You can look at some locations of interest. Peaks with low FDR (q value) or high fold enrichment may be worth checking out. We find these peaks by numerically sorting the results in broadPeak by column “score” (the 5th column). Or check your favourite gene.
Potentially interesting locations to view (MACS peaks).
Let’s sort the broadPeak file using the score column to find the peaks with the strongest signal
sort -k5,5rn neuroGM23338_macs3_rep1_peaks.broadPeak | head
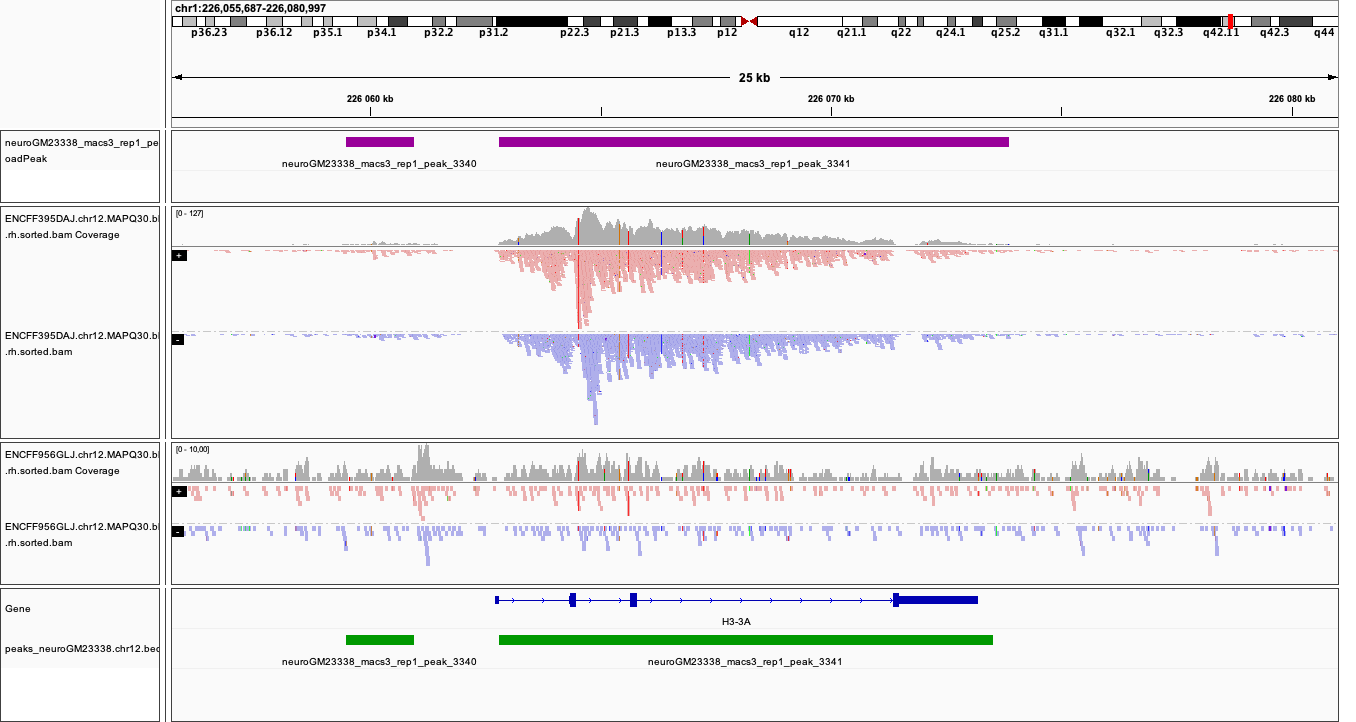
chr1 226062814 226073870 neuroGM23338_macs3_rep1_peak_3341 518 . 13.0292 54.709 51.8498
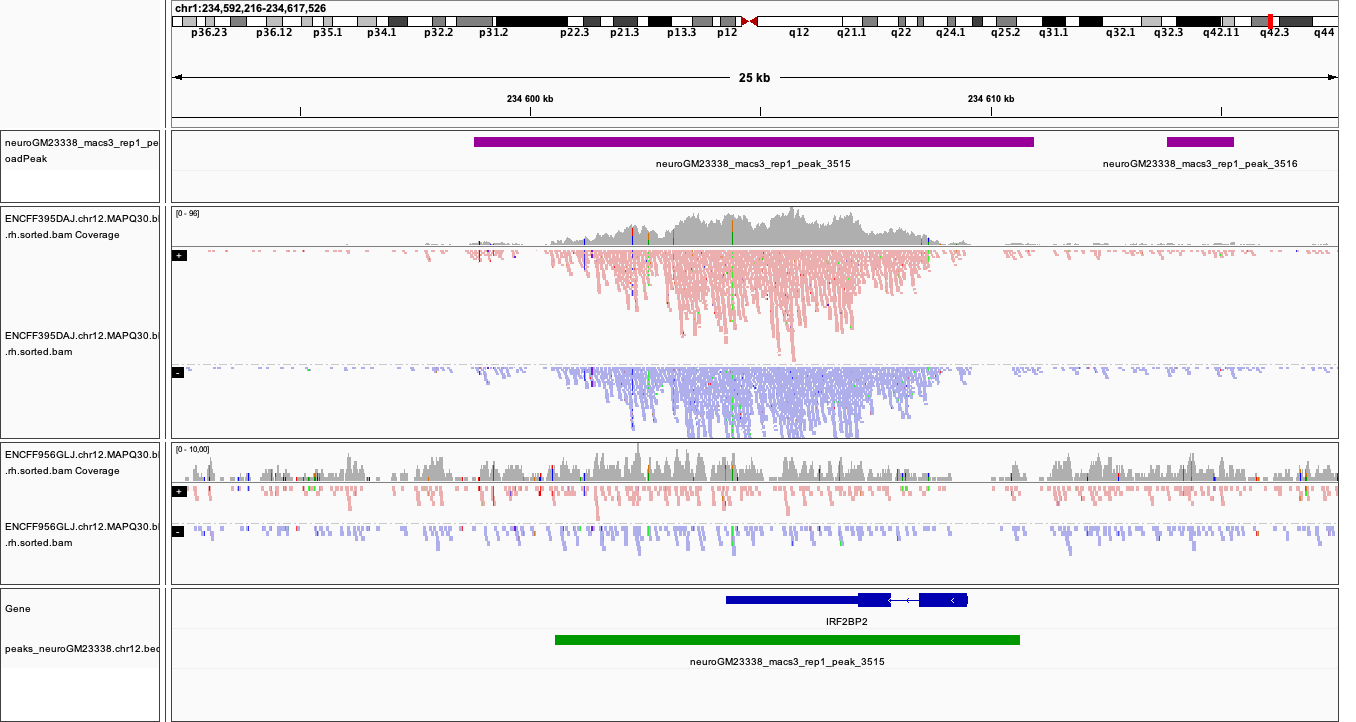
chr1 234598782 234610959 neuroGM23338_macs3_rep1_peak_3515 513 . 12.139 54.2276 51.3993
chr2 101698297 101748719 neuroGM23338_macs3_rep1_peak_5147 462 . 13.375 49.0116 46.2392
chr2 47158830 47176361 neuroGM23338_macs3_rep1_peak_4479 449 . 12.5555 47.6423 44.96
chr1 204403186 204412701 neuroGM23338_macs3_rep1_peak_2999 431 . 10.9654 45.8227 43.1784
chr1 220527779 220538029 neuroGM23338_macs3_rep1_peak_3239 414 . 11.5724 44.1237 41.4922
chr1 244049535 244060201 neuroGM23338_macs3_rep1_peak_3688 403 . 10.2749 42.9757 40.397
chr2 54970096 55050304 neuroGM23338_macs3_rep1_peak_4577 401 . 11.3781 42.7092 40.1167
chr2 5692693 5703228 neuroGM23338_macs3_rep1_peak_3837 399 . 10.6746 42.502 39.9488
chr1 150568640 150579241 neuroGM23338_macs3_rep1_peak_2121 394 . 11.0116 42.0139 39.4717
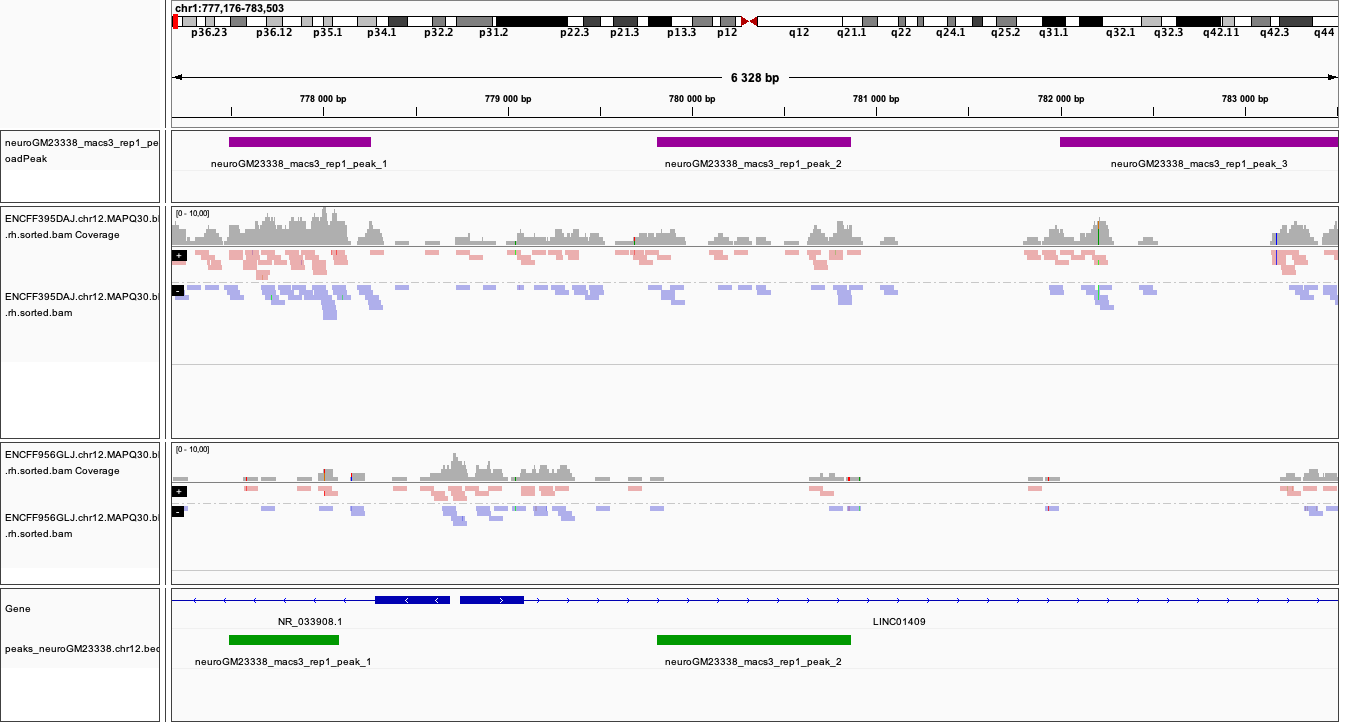
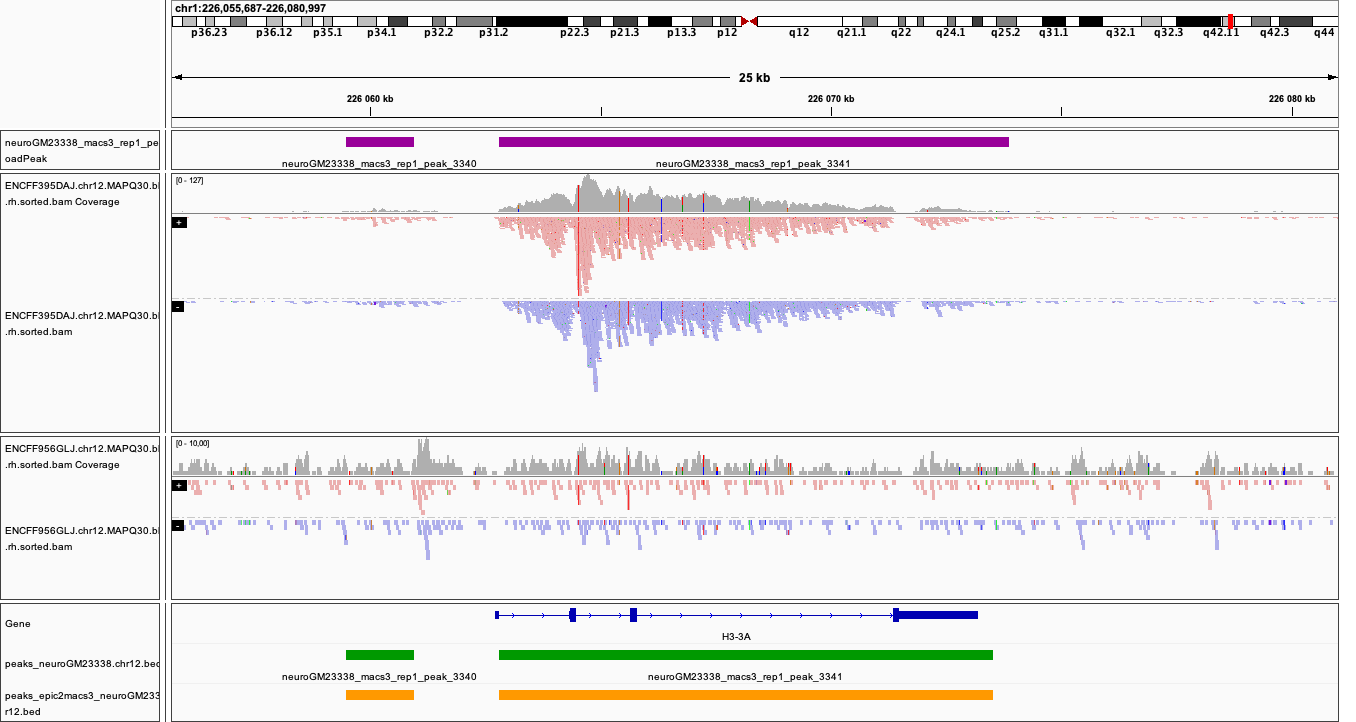
Below you see IGV visualisations of the following regions (top two peaks and one of the bottom rank):
chr1:226,055,687-226,080,997
chr1:234,592,216-234,617,526
chr1:777,176-783,503
IGV settings for this visualiation (right click on the left hand side panel with track names): Group alignments (by read strand); Colour alignments (by read strand); Squished.
Regions detected by MACS3 are the topmost purple track, two bam files are ChIP and input (with their pileups calculated by IGV), and the bottom panel are gene models and, finally the regions reproducible between both replicates in green.
Please note the length of these detected domains.
|
|
|
Broad peak calling using epic2
epic2 is an ultraperformant reimplementation of SICER, an algorithm developed especially for detection of broad marks. It focuses on speed, low memory overhead and ease of use. It also contains a reimplementation of the SICER-df scripts for differential enrichment and a script to create many kinds of bigwigs from your data. In this tutorial we will use it to detect domains in the same data as we used earlier for MACS. At the end we will compare the results.
Here again we use a prepared conda environment. Newer versions of Pysam seem to throw errors when used with epic2. For details please consult Dependencies.
mkdir ../../epic2
cd ../../epic2
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338/ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338/ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam.bai
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338//ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338//ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam.bai
conda activate /sw/courses/epigenomics/software/conda/epic_2b
epic2 --treatment ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam \
--control ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam \
-fdr 0.05 --effective-genome-fraction 0.95 \
--chromsizes /sw/courses/epigenomics/broad_peaks2/annot/hg38_chr12.chromsizes \
--guess-bampe --output neuroGM23338_epic2_rep1_peaks
The result is a coordinate sorted text file:
head neuroGM23338_epic2_rep1_peaks
#Chromosome Start End PValue Score Strand ChIPCount InputCount FDR log2FoldChange
chr1 777400 778199 1.525521715195486e-17 302.351431362403 . 28 4 3.6469084588658344e-17 3.02351431362403
chr1 821600 822599 9.82375064925635e-15 309.0628509482567 . 22 3 2.149649196967778e-14 3.090628509482567
chr1 823400 826599 1.912606813336636e-50 258.96177870938703 . 114 22 7.16207410279741e-50 2.58961778709387
chr1 828200 830399 5.824989404621774e-19 201.13395996779278 . 59 17 1.4498521203360967e-18 2.0113395996779277
chr1 831400 833599 2.388427396703667e-14 151.73289262869903 . 69 28 5.17334134771682e-14 1.5173289262869905
chr1 880600 885799 2.569723734705377e-37 153.80874864537878 . 195 78 8.520333236155976e-37 1.538087486453788
chr1 886600 890199 1.481460204837024e-22 165.05622157122005 . 100 37 3.9835062883707675e-22 1.6505622157122006
chr1 925800 926999 8.491192455372113e-17 280.11218922875815 . 30 5 1.9845609170824598e-16 2.801121892287582
chr1 957000 959199 4.1356938759907084e-64 337.1437617044337 . 98 11 1.688095302270476e-63 3.3714376170443368
The meaning of the columns:
- PValue
Poisson-computed PValue based on the number of ChIP count vs. library-size normalized Input count in the region
- Score
Log2FC * 100 (capped at 1000). Regions with a larger relative ChIP vs. Input count will show as darker in the UCSC genome browser
- ChIPCount
The number of ChIP counts in the region (also including counts from windows with a count below the cutoff)
- InputCount
The number of Input counts in the region
- FDR
Benjamini-Hochberg correction of the p-values
- log2FoldChange
Log2 of the region ChIP count vs. the library-size corrected region Input count
How many domains were found? (the first line is a header)
wc -l neuroGM23338_epic2_rep1_peaks
5242 neuroGM23338.rep1.epic2
How many domains reproducible between replicates?
Select reproducible peaks (epic2).
mkdir peaks
cd peaks
#link the files
ln -s /sw/courses/epigenomics/broad_peaks2021/results/epic2/neuroGM23338/neuroGM23338.rep1.epic2
ln -s /sw/courses/epigenomics/broad_peaks2021/results/epic2/neuroGM23338/neuroGM23338.rep2.epic2
#make bed
cut -f 1-3 neuroGM23338.rep1.epic2 >neuroGM23338_epic2_rep1_peaks.bed
cut -f 1-3 neuroGM23338.rep2.epic2 >neuroGM23338_epic2_rep2_peaks.bed
#intersect bed files
module load bioinfo-tools #if necessary
module load BEDTools/2.29.2
bedtools intersect -a neuroGM23338_epic2_rep1_peaks.bed -b neuroGM23338_epic2_rep2_peaks.bed -f 0.50 -r > peaks_epic2_neuroGM23338.chr12.bed
#how many peaks which overlap?
wc -l peaks_epic2_neuroGM23338.chr12.bed
2692 peaks_epic2_neuroGM23338.chr12.bed
How about the overlap between different methods?
Compare MACS3 and epic2.
(please make sure the relative path to macs3 results is correct in the command below)
#intersect bed files
module load bioinfo-tools #if necessary
module load BEDTools/2.29.2
bedtools intersect -a peaks_epic2_neuroGM23338.chr12.bed -b ../../macs3/peaks/peaks_macs3_neuroGM23338.chr12.bed \
-f 0.50 -r > peaks_epic2macs3_neuroGM23338.chr12.bed
#how many peaks which overlap?
wc -l peaks_epic2macs3_neuroGM23338.chr12.bed
1629 peaks_epic2macs3_neuroGM23338.chr12.bed
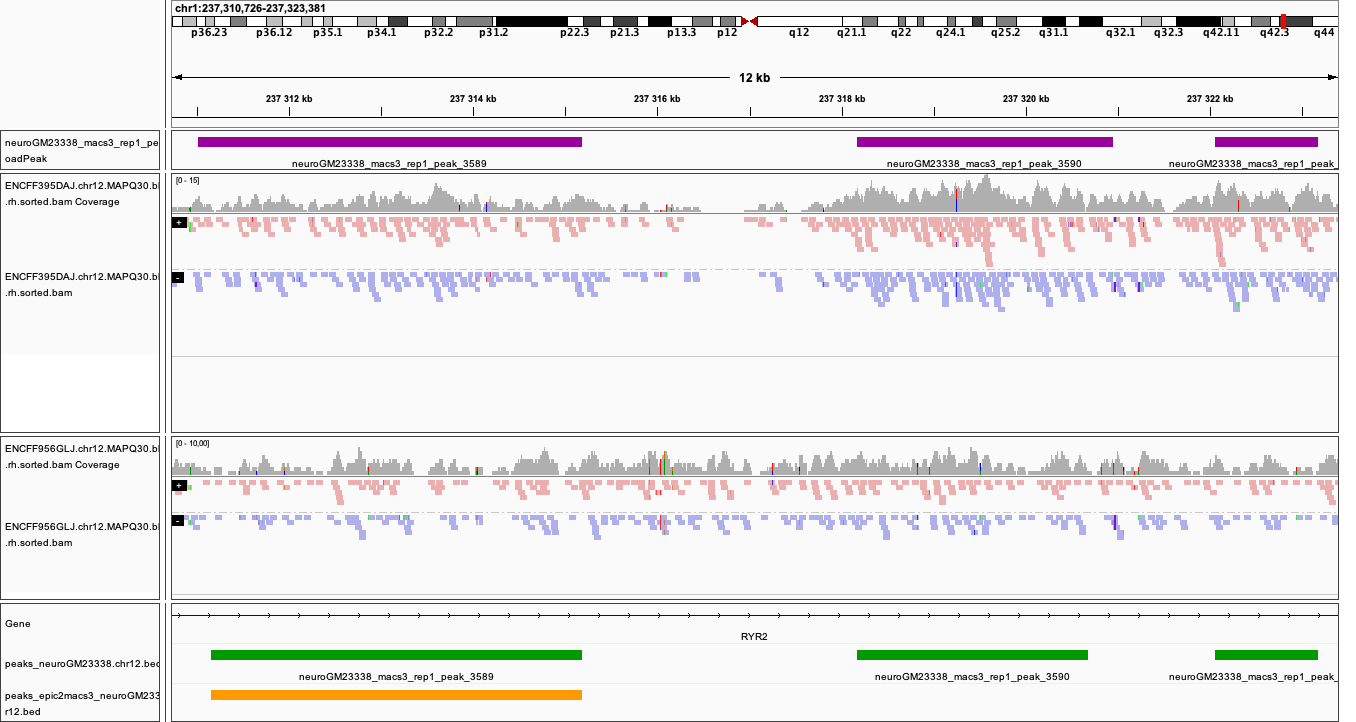
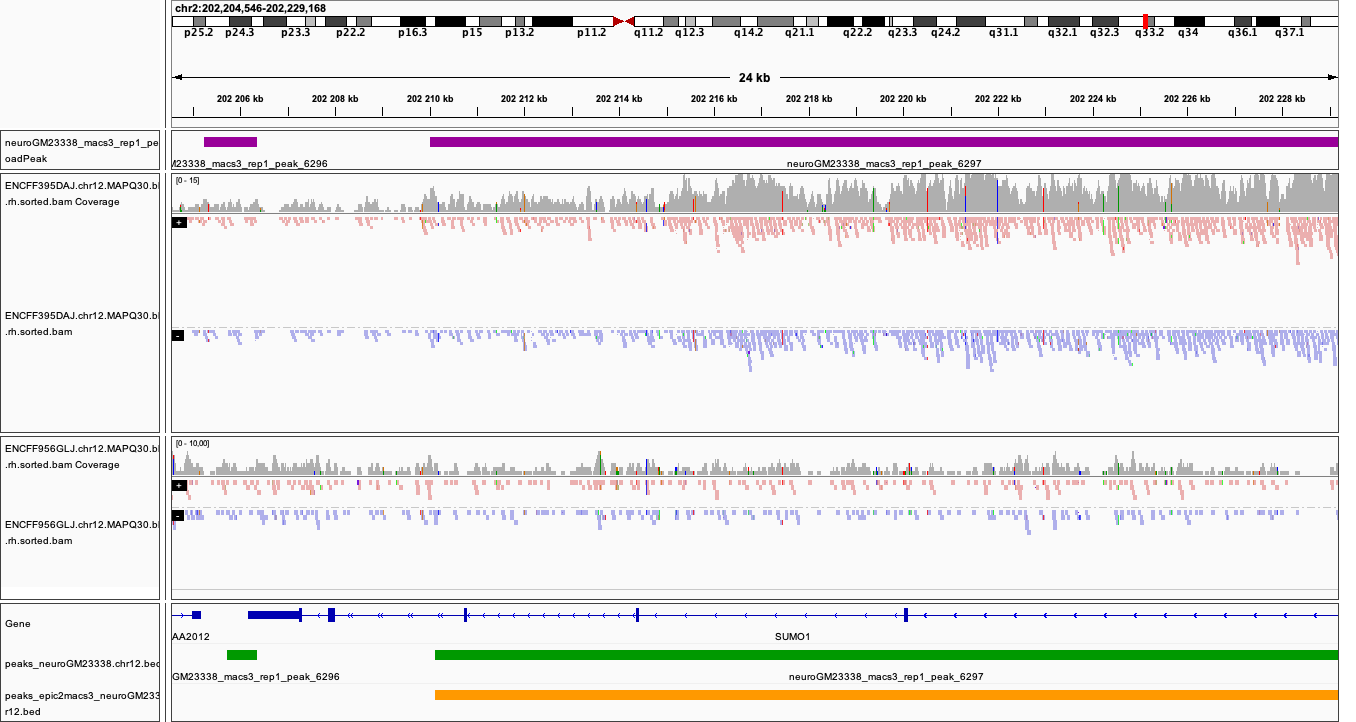
You can visualise the peaks as for MACS in the earlier section. Below are plots of some of the locations as in the MACS, with peaks detected by both epic2 and MACS marked in orange.
|
|
|
Locations plotted.
chr1:226,055,687-226,080,997
chr1:237,310,726-237,323,381
chr2:202,204,546-202,229,168
You can now deactivate the conda environment you’ve been working in:
conda deactivate
Alternative approach: window-based enrichment analysis (csaw)
This is an alternative workflow for detection of differential binding / occupancy in ChIP-seq data. In this mode, in contrast to working with reads counted within peaks detected in a peak calling step (as in the earlier example with DiffBind), this approach uses a sliding window to count reads across the genome. Each window is then tested for significant differences between libraries from different conditions, using the methods in the edgeR package. This package also offers an FDR control strategy more appropriate for ChIP-seq experiments than simple BH adjustment.
csaw can also be used to detect peaks - if the sample to compare to is input.
As this method is agnostic to signal structure, it requires careful choice of strategies for filtering and normalisation. Here, we show a very simple workflow. More details can be found in the Csaw User Guide.
Note
This exercise was tested on Rackham using pre-installed R libraries. Local installation of recommended R packages may require additional software dependecies. Please see Dependencies for details.
Requirements Remote (Uppmax)
The software is configured, i.e. the correct R version is loaded via the module system and required libraries are preinstalled.
To prepare the files, assuming you are in ~/broad_peaks/analysis:
mkdir csaw
cd csaw
mkdir bam
ln -s /proj/epi2025/broad_peaks/data/neuron_GM23338/* bam
module load R_packages/4.0.4
The remaining part of the exercise is performed in R.
Sort out the working directory and file paths:
setwd("/path/to/workdir")
dir.data = "/path/to/desired/location/bam"
#for example when in broad_peaks/csaw
dir.data = "./bam"
k79_1=file.path(dir.data,"ENCFF395DAJ.chr12.MAPQ30.blcklst.rh.sorted.bam")
input_1=file.path(dir.data,"ENCFF956GLJ.chr12.MAPQ30.blcklst.rh.sorted.bam")
k79_2=file.path(dir.data,"ENCFF806KRA.chr12.MAPQ30.blcklst.rh.sorted.bam")
input_2=file.path(dir.data,"ENCFF687LIL.chr12.MAPQ30.blcklst.rh.sorted.bam")
bam.files <- c(k79_1,k79_2,input_1,input_2)
Hint
Setting the paths in R
To find the path to your current location type pwd in the terminal. You can use this path in R like this:
setwd("/path/to/where_you_are")
All the paths will be then relative to /path/to/where_you_are.
You can also find it directly from R using getwd:
> getwd()
[1] "/crex/course_data/epigenomics/broad_peaks2021/results/csaw"
Read in the data:
library(csaw)
pe.param <- readParam(max.frag=400, pe="both")
data <- windowCounts(bam.files, width=100, param=pe.param)
ChIP experiments with paired-end sequencing are accomodated by setting pe="both" in the
param object supplied to windowCounts. Read extension is not required as the genomic interval
spanned by the originating fragment is explicitly defined as that between the 5′positions of
the paired reads. By default, only proper pairs are used in which the two paired reads are
on the same chromosome, face inward and are no more than max.frag apart (we use the default value 500, which is suitable for most libraries).
width specifies the width of the window when counting the fragments.
How many valid windows do we have?:
data$totals
[1] 3666329 5635840 4436456 16125939
data
> data
class: RangedSummarizedExperiment
dim: 8808414 4
metadata(6): spacing width ... param final.ext
assays(1): counts
rownames: NULL
rowData names(0):
colnames: NULL
colData names(4): bam.files totals ext rlen
You will identify the enrichment windows by performing a differential occupancy analysis between ChIP and input samples.
Information on the contrast to test:
grouping <- factor(c('chip', 'chip', 'input', 'input'))
design <- model.matrix(~0 + grouping)
colnames(design) <- levels(grouping)
library(edgeR)
contrast <- makeContrasts(chip - input, levels=design)
contrast
> contrast
Contrasts
Levels chip - input
chip 1
input -1
Next, you need to filter out uninformative windows with low signal prior to further analysis. Selection of appropriate filtering strategy and cutoff is key to a successful detection of differential occupancy events, and is data dependent. Filtering is valid so long as it is independent of the test statistic under the null hypothesis. One possible approach involves choosing a filter threshold based on the fold change over the level of non-specific enrichment (background). The degree of background enrichment is estimated by counting reads in large bins across the genome.
The function filterWindowsGlobal returns the increase in the abundance of
each window over the global background.
Windows are filtered by setting some minimum threshold on this increase. Here, a fold change of 3 is necessary for a window to be considered as containing a binding site. This and other filtering procedures are described in detail in
csaw user guide
. We use the “By global enrichment” strategy.
In this example, you estimate the global background using ChIP samples only. You can do it using the entire dataset including inputs of course.
bam.files_chip <- c(k79_1,k79_2)
bin.size <- 2000L
binned.ip <- windowCounts(bam.files_chip, bin=TRUE, width=bin.size, param=pe.param)
data.ip=data[,1:2]
filter.stat <- filterWindowsGlobal(data.ip, background=binned.ip)
keep <- filter.stat$filter > log2(3)
data.filt <- data[keep,]
To examine how many windows passed the filtering:
summary(keep)
Mode FALSE TRUE
logical 7466311 731112
To normalise the data for different library sizes you need to calculate normalisation factors based on large bins:
binned <- windowCounts(bam.files, bin=TRUE, width=10000, param=pe.param)
data.filt <- normFactors(binned, se.out=data.filt)
data.filt$norm.factors
## [1] 0.6094691 0.6654708 1.5651132 1.5753374
Detection of DB (differentially bound) windows (in our case, the occupancy sites, as we test for differences in ChIP vs. input):
data.filt.calc <- asDGEList(data.filt)
data.filt.calc <- estimateDisp(data.filt.calc, design)
fit <- glmQLFit(data.filt.calc, design, robust=TRUE)
results <- glmQLFTest(fit, contrast=contrast)
You can inspect the raw results:
head(results$table)
logFC logCPM F PValue
1 4.397419 0.1113531 39.71585 1.054723e-07
2 3.957880 0.1781093 36.54985 2.550295e-07
3 4.079911 0.2803444 42.31232 5.243527e-08
4 3.920461 0.4808799 47.20246 1.487789e-08
5 4.410081 0.5664205 59.33251 8.606713e-10
6 5.026440 0.6390274 69.96147 9.239046e-11
The following steps will calculate the FDR for each peak, merge peaks within 1 kb and calculate the FDR for resulting composite peaks.
merged <- mergeWindows(rowRanges(data.filt), tol=1000L)
table.combined <- combineTests(merged$id, results$table)
Short inspection of the results:
head(table.combined)
DataFrame with 6 rows and 8 columns
num.tests num.up.logFC num.down.logFC PValue FDR direction
<integer> <integer> <integer> <numeric> <numeric> <character>
1 12 12 0 1.10869e-09 6.83746e-09 up
2 9 9 0 5.54991e-06 1.53993e-05 up
3 54 54 0 1.28669e-10 9.16836e-10 up
4 29 29 0 3.25906e-08 1.59822e-07 up
5 36 36 0 3.14755e-07 1.26463e-06 up
6 1 1 0 1.06514e-05 2.62040e-05 up
rep.test rep.logFC
<integer> <numeric>
1 6 5.02644
2 14 4.11686
3 61 4.42842
4 85 3.70258
5 113 3.45417
6 141 3.45264
How many regions are up (i.e. enriched in chip compared to input)?
is.sig.region <- table.combined$FDR <= 0.1
table(table.combined$direction[is.sig.region])
up
8116
Does this make sense? How does it compare to results obtained from MACS and epic2 runs?
You can now annotate the results:
library(org.Hs.eg.db)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
anno <- detailRanges(merged$region, txdb=TxDb.Hsapiens.UCSC.hg38.knownGene,
orgdb=org.Hs.eg.db, promoter=c(3000, 1000), dist=5000)
merged$region$overlap <- anno$overlap
merged$region$left <- anno$left
merged$region$right <- anno$right
all.results <- data.frame(as.data.frame(merged$region)[,1:3], table.combined, anno)
sig=all.results[all.results$FDR<0.05,]
all.results <- all.results[order(all.results$PValue),]
head(all.results)
filename="k79me2_neuroGM_csaw.txt"
write.table(all.results,filename,sep="\t",quote=FALSE,row.names=FALSE)
Let’s inspect the results:
head(all.results)
seqnames start end num.tests num.up.logFC num.down.logFC
3799 chr1 226062751 226073900 213 213 0
870 chr1 35176951 35193200 319 319 0
5154 chr2 47157601 47178000 344 344 0
4233 chr1 244835751 244867200 617 617 0
4003 chr1 234598601 234610800 211 211 0
2608 chr1 160363601 160374200 205 205 0
PValue FDR direction rep.test rep.logFC
3799 2.124641e-23 1.306798e-19 up 351746 6.758585
870 5.493232e-23 1.306798e-19 up 92252 7.177477
5154 6.358299e-23 1.306798e-19 up 459830 6.833969
4233 6.750822e-23 1.306798e-19 up 390369 6.430975
4003 8.050751e-23 1.306798e-19 up 369849 6.761304
2608 1.305657e-22 1.560297e-19 up 253600 6.381518
overlap left right
3799 H3-3A:+:PE,H3P6:+:PE H3P6:+:657
870 RNVU1-18:-:I,SFPQ:-:PE SFPQ:-:477
5154 STPG4:-:PE,CALM2:-:PE STPG4:-:2293
4233 COX20:+:PE,HNRNPU:-:PE
4003 IRF2BP2:-:PE
2608 NHLH1:+:PE NCSTN:+:4649
To compare with peaks detected by MACS it is convenient to save the results in BED format:
sig.up=sig[sig$direction=="up",]
starts=sig.up[,2]-1
sig.up[,2]=starts
sig_bed=sig.up[,c(1,2,3)]
filename="k79me2_neuroGM_csaw_peaks.bed"
write.table(sig_bed,filename,sep="\t",col.names=FALSE,quote=FALSE,row.names=FALSE)
You can now load the bed file to IGV along with the appropriate broad.Peak file and zoom in to your favourite location on chromosomes 1 and 2.
Below is the IGV snapshot of top peak, this time with csaw peaks added in light blue.
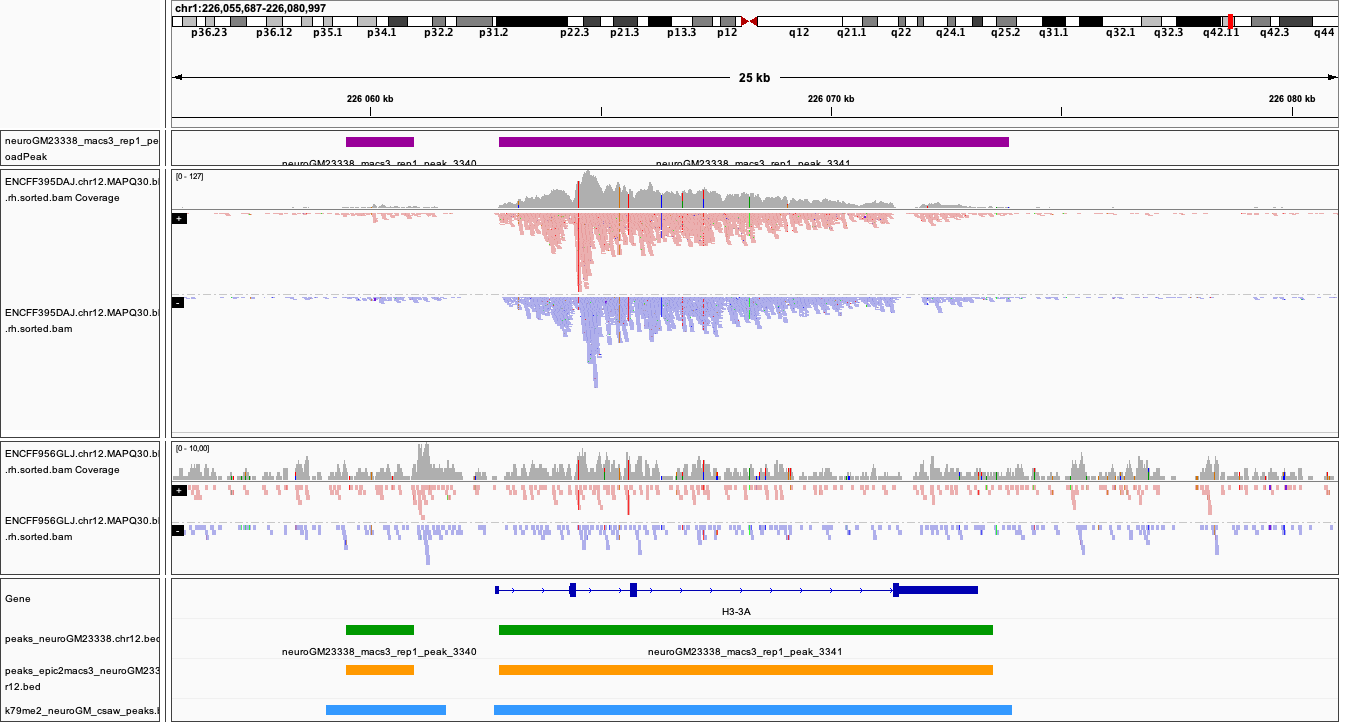
|
As you can see the regions with strong signal (high enrichment in ChIP over input) are detected by all methods tested. What about the sites with weak signal?
In this tutorial we have worked with good quality data which was sequenced to a recommended depth. All three methods tested in this tutorial perform well is such scenario. However, their preformace deteriorates with decreasing sequening depth (less data to rely on) and decreasing quality of the sample preparation (more noise).
sessionInfo()
Random number generation:
RNG: Mersenne-Twister
Normal: Inversion
Sample: Rejection
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] TxDb.Hsapiens.UCSC.hg38.knownGene_3.10.0
[2] GenomicFeatures_1.42.3
[3] org.Hs.eg.db_3.12.0
[4] AnnotationDbi_1.52.0
[5] edgeR_3.32.1
[6] limma_3.46.0
[7] csaw_1.24.3
[8] SummarizedExperiment_1.20.0
[9] Biobase_2.50.0
[10] MatrixGenerics_1.2.1
[11] matrixStats_0.58.0
[12] GenomicRanges_1.42.0
[13] GenomeInfoDb_1.26.7
[14] IRanges_2.24.1
[15] S4Vectors_0.28.1
[16] BiocGenerics_0.36.0
loaded via a namespace (and not attached):
[1] locfit_1.5-9.4 Rcpp_1.0.6 lattice_0.20-41
[4] Rsamtools_2.6.0 prettyunits_1.1.1 Biostrings_2.58.0
[7] assertthat_0.2.1 utf8_1.2.1 BiocFileCache_1.14.0
[10] R6_2.5.0 RSQLite_2.2.6 httr_1.4.2
[13] pillar_1.6.0 zlibbioc_1.36.0 rlang_0.4.10
[16] progress_1.2.2 curl_4.3 rstudioapi_0.13
[19] blob_1.2.1 Matrix_1.3-2 splines_4.0.4
[22] statmod_1.4.35 BiocParallel_1.24.1 stringr_1.4.0
[25] RCurl_1.98-1.3 bit_4.0.4 biomaRt_2.46.3
[28] DelayedArray_0.16.3 compiler_4.0.4 rtracklayer_1.50.0
[31] pkgconfig_2.0.3 askpass_1.1 openssl_1.4.3
[34] tidyselect_1.1.0 tibble_3.1.1 GenomeInfoDbData_1.2.4
[37] XML_3.99-0.6 fansi_0.4.2 crayon_1.4.1
[40] dplyr_1.0.5 dbplyr_2.1.1 GenomicAlignments_1.26.0
[43] bitops_1.0-6 rappdirs_0.3.3 grid_4.0.4
[46] lifecycle_1.0.0 DBI_1.1.1 magrittr_2.0.1
[49] stringi_1.5.3 cachem_1.0.4 XVector_0.30.0
[52] xml2_1.3.2 ellipsis_0.3.1 vctrs_0.3.7
[55] generics_0.1.0 tools_4.0.4 bit64_4.0.5
[58] glue_1.4.2 purrr_0.3.4 hms_1.0.0
[61] fastmap_1.1.0 memoise_2.0.0