Differential Accessibility in ATAC-seq
- Date:
2025-09-16
Learning outcomes
to interrogate GC bias in ATAC-seq data
to select appropriate scaling normalisation of ATAC-seq data
to detect differentially accessible regions using
edgeR
Note
We continue working with data from (Tsao et al. 2022). We will use the count table derived from non subset data (already prepared).
Introduction
In this tutorial we use an R / Bioconductor packages edgeR (Robinson
and Oshlack 2010), (Chen, Lun, and Smyth 2016), EDASeq (Risso et al.
2011) and cqn (Hansen, Irizarry, and WU 2012) to perform
normalisation and analysis of differential accessibility in ATAC-seq
data.
Data & Methods
We will build upon the main lab ATACseq data analysis:
we will interrogate GC bias in peaks and in adjacent non-overlapping genomic bins;
we will use the counts table encompassing complete data for differential accessibility analysis;
Setting Up
You can continue working in the directory atacseq/analysis/DA.
This directory contains count tables derived from summarising of non-subset data to merged peaks called by genrich in the joint mode. We
will use file
AB_Batf_KO_invivo.genrich_joint.merged_peaks.featureCounts.
We can link additional files:
mkdir bam_dir
find /proj/epi2025/atacseq_proc/data_proc/ -name \*.ba* -exec ln -vs "{}" bam_dir/ ';'
We access the R environment via:
module load R_packages/4.3.1
We activate R console upon typing R in the terminal.
We begin by loading necessary libraries:
library(tidyverse)
library(dplyr)
library(kableExtra)
library(ggplot2)
library(wesanderson)
library(GenomicRanges)
library(Hmisc)
library(Biostrings)
library(regioneR)
library(bamsignals)
require(MASS)
library(BSgenome.Mmusculus.UCSC.mm39)
library(edgeR)
library(limma)
library(SummarizedExperiment)
library(EDASeq)
library(cqn)
workdir=getwd()
To set working directory to your desired path you can use these commands:
workdir="/path/to/workdir"
workdir=setwd()
Note
We take advantage of the module system on Rackham in this tutorial. The
code was tested under R 4.3.1 The lab was developed under different
R version, as stated in session info.
Data
We can now load data. We will subset the count table to only contain the peaks on assembled chromosomes.
count_table_fname="AB_Batf_KO_invivo.genrich_joint.merged_peaks.featureCounts"
cnt_table_pth=file.path(file.path(workdir,"counts"),count_table_fname)
cnt_table=read.table(cnt_table_pth, sep="\t", header=TRUE, blank.lines.skip=TRUE)
rownames(cnt_table)=cnt_table$Geneid
rownames(cnt_table)=c(gsub("AB_Batf_KO_invivo.genrich_joint.","",rownames(cnt_table)))
colnames(cnt_table)=c(colnames(cnt_table)[1:6],gsub(".filt.bam","",colnames(cnt_table)[7:10]))
colnames(cnt_table)[7:10]=c("B1_WT_Batf-floxed","B2_WT_Batf-floxed","A1_Batf_cKO","A2_Batf_cKO")
#remove peaks not on the assembled chromosomes
cnt_table_chr=cnt_table|>
dplyr::filter(Chr%in%c(1:19) | Chr%in%c("X","Y"))
reads.peak=cnt_table_chr[,c(7:10)]
head(reads.peak)
## B1_WT_Batf-floxed B2_WT_Batf-floxed A1_Batf_cKO A2_Batf_cKO
## merged_peaks_1 299 238 325 330
## merged_peaks_2 106 83 162 174
## merged_peaks_3 19 24 25 21
## merged_peaks_4 27 31 40 29
## merged_peaks_5 114 101 65 151
## merged_peaks_6 129 137 120 204
All peaks: n = 65027.
Peaks on assembled chromosomes: n = 64879. These peaks will be used for further analysis.
GC Bias
GC Bias in Genomic Bins
To ivestigate the GC bias in adjacent genomic bins (background), we
start with creating the GRanges object holding the tiled genome
intervals. We will do it for one chromosome only (chr1), to save compute
time.
chr.lengths = seqlengths(Mmusculus)[1:21]
chr.lengths.chr1=chr.lengths[1]
#tiles
tiles_chr1=GenomicRanges::tileGenome(chr.lengths.chr1,tilewidth=5000, cut.last.tile.in.chrom=TRUE)
#sequence
tileSeqs=BSgenome::getSeq(Mmusculus,tiles_chr1)
#GCcontent
gcContentTiles=Biostrings::letterFrequency(tileSeqs, "GC",as.prob=TRUE)[,1]
mcols(tiles_chr1)$gc=gcContentTiles
We need to tweak chromosome names to match the genome reference used for read mapping:
# tiles_chr1
# remove chr from granges obj
seqlevels(tiles_chr1)=gsub("chr","",seqlevels(tiles_chr1))
We can now count reads in all bam files in the data set, and plot them.
bam_dir=file.path(workdir,"bam_dir")
bam_fnames=list.files(bam_dir,pattern = "\\.bam$",)
par(mfrow = c(2, length(bam_fnames)/2 ) )
for (bam_fname in bam_fnames){
bam_path=file.path(bam_dir,bam_fname)
tiles_bam=tiles_chr1
tiled_counts=bamCount(bam_path, tiles_bam, verbose=FALSE)
mcols(tiles_bam)$readcount=tiled_counts
smoothScatter(tiles_bam$gc, log2(tiles_bam$readcount+1),
main=paste("Logcounts vs GC in bins",bam_fname,sep="\n"), ylab="log(counts+1)", xlab="GC content")
}
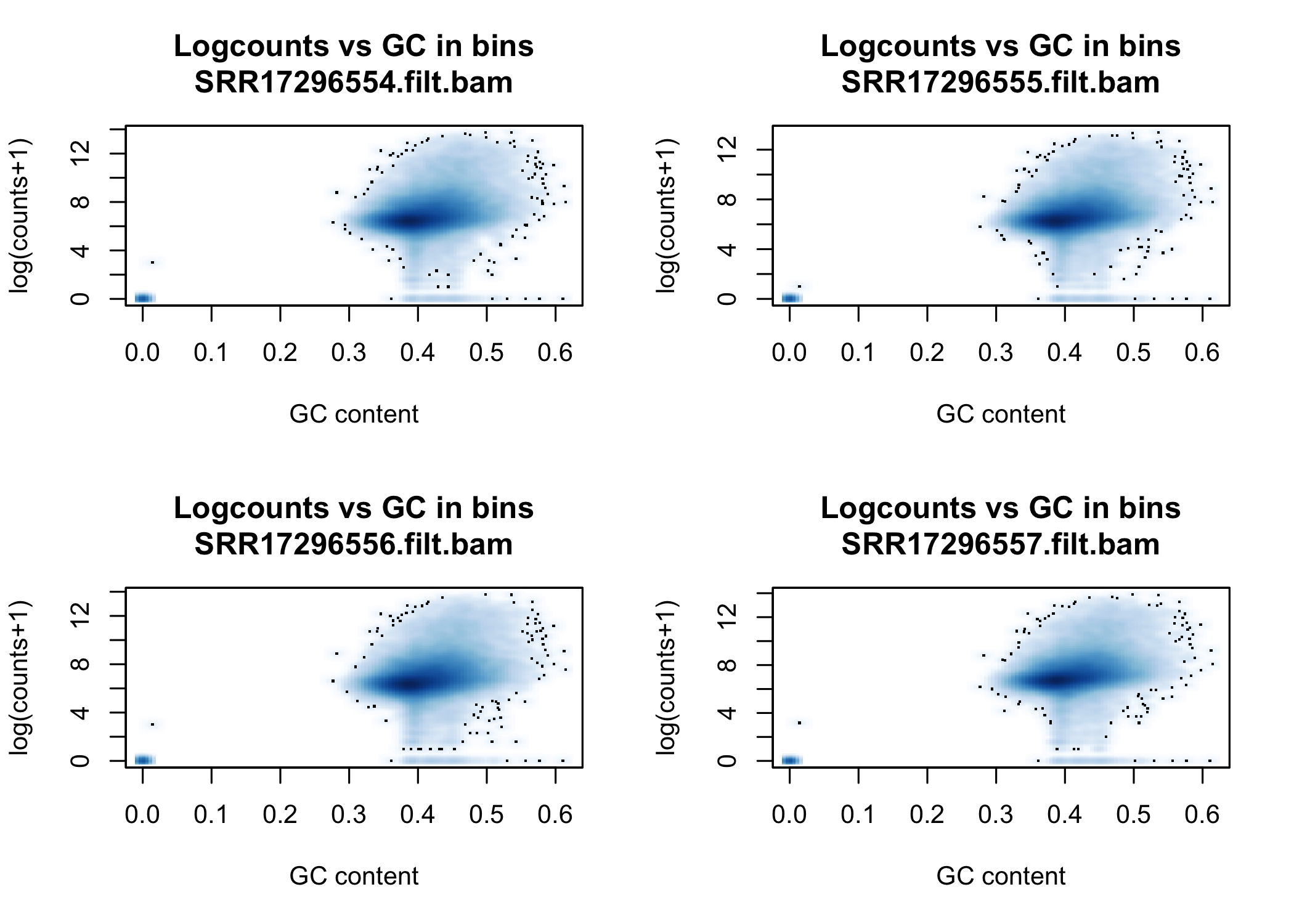
We can see that the signal of logcounts vs GC content looks very similar in all libraries.
GC Bias in Peaks
To ivestigate the GC bias in peaks (signal), we start with creating the
GRanges object holding the peak intervals.
We need to prefix the chromosome name by “chr” (per UCSC convention) in
the first step to be able to use a BSgenome object from the
Bioconductor package BSgenome.Mmusculus.UCSC.mm39. Please note this
only works with assembled chromosomes; the non-assembled contigs follow
different naming conventions in Ensembl (the source of the reference
assembly for read mapping) and UCSC (the source of BSgenome package).
peaks_gr=GRanges(seqnames=paste0("chr",cnt_table_chr$Chr), ranges=IRanges(cnt_table_chr$Start, cnt_table_chr$End), strand="*", mcols=data.frame(peakID=rownames(cnt_table_chr)))
We now prepare data with GC content of the peak regions for GC-aware normalisation.
peakSeqs=BSgenome::getSeq(Mmusculus,peaks_gr)
gcContentPeaks=Biostrings::letterFrequency(peakSeqs, "GC",as.prob=TRUE)[,1]
#divide into 20 bins by GC content
gcGroups=Hmisc::cut2(gcContentPeaks, g=20)
mcols(peaks_gr)$gc=gcContentPeaks
mcols(peaks_gr)$gc_group=gcGroups
peaks_gr
## GRanges object with 64879 ranges and 3 metadata columns:
## seqnames ranges strand | mcols.peakID gc
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] chr1 3050939-3052959 * | merged_peaks_1 0.392875
## [2] chr1 3053048-3054634 * | merged_peaks_2 0.379962
## [3] chr1 3054861-3055532 * | merged_peaks_3 0.345238
## [4] chr1 3057260-3057785 * | merged_peaks_4 0.376426
## [5] chr1 3059375-3061360 * | merged_peaks_5 0.402316
## ... ... ... ... . ... ...
## [64875] chrY 90814281-90815165 * | merged_peaks_64875 0.505085
## [64876] chrY 90815739-90816707 * | merged_peaks_64876 0.430341
## [64877] chrY 90818033-90819321 * | merged_peaks_64877 0.493406
## [64878] chrY 90819900-90820364 * | merged_peaks_64878 0.369892
## [64879] chrY 90821996-90824312 * | merged_peaks_64879 0.469141
## gc_group
## <factor>
## [1] [0.234,0.396)
## [2] [0.234,0.396)
## [3] [0.234,0.396)
## [4] [0.234,0.396)
## [5] [0.396,0.417)
## ... ...
## [64875] [0.505,0.514)
## [64876] [0.417,0.431)
## [64877] [0.487,0.496)
## [64878] [0.234,0.396)
## [64879] [0.461,0.470)
## -------
## seqinfo: 21 sequences from an unspecified genome; no seqlengths
Figure below shows that the accessibility measure of a particular genomic region is associated with its GC content. In this data set, the curves are almost identical for all samples, indicating no difference in GC bias between samples.
However, in some cases the slope and shape of the curves may differ between samples, which indicates that GC content effects are sample–specific and can therefore bias between–sample comparisons.
We start by creating a data frame with gc contents and read count in
each peak in each sample as well as perform lowess (locally weighted
scatterplot smoothing) regression to fit the trend:
lowListGC = list()
for(kk in 1:ncol(reads.peak)){
set.seed(kk)
lowListGC[[kk]] = lowess(x=gcContentPeaks, y=log1p(reads.peak[,kk]), f=1/10)
}
names(lowListGC)=colnames(reads.peak)
dfList = list()
for(ss in 1:length(lowListGC)){
oox = order(lowListGC[[ss]]$x)
dfList[[ss]] = data.frame(x=lowListGC[[ss]]$x[oox], y=lowListGC[[ss]]$y[oox], sample=names(lowListGC)[[ss]])
}
dfAll = do.call(rbind, dfList)
dfAll$sample = factor(dfAll$sample)
We can now plot the relationship of logcounts vs GC content:
plotGCHex <- function(gr, counts){
counts2 <- counts
df <- as_tibble(cbind(counts2,gc=mcols(gr)$gc))
df <- gather(df, sample, value, -gc)
ggplot(data=df, aes(x=gc, y=log(value+1)) ) +
ylab("log(count + 1)") + xlab("GC-content") +
geom_hex(bins = 50) + theme_bw()
}
plot_GC_bias=plotGCHex(peaks_gr, rowMeans(reads.peak)) +
theme(axis.title = element_text(size=16)) +
labs(fill="Nr. of peaks") +
geom_line(aes(x=x, y=y, group=sample, color=sample), data=dfAll, linewidth=1) +
scale_color_discrete()

Differential Accessibility
We can define experimental groups:
groups=factor(c(rep("ctrl",2),rep("KO_Batf",2)))
groups
## [1] ctrl ctrl KO_Batf KO_Batf
## Levels: ctrl KO_Batf
design=model.matrix(~groups)
rownames(design)=colnames(reads.peak)
design
## (Intercept) groupsKO_Batf
## B1_WT_Batf-floxed 1 0
## B2_WT_Batf-floxed 1 0
## A1_Batf_cKO 1 1
## A2_Batf_cKO 1 1
## attr(,"assign")
## [1] 0 1
## attr(,"contrasts")
## attr(,"contrasts")$groups
## [1] "contr.treatment"
We’ll detect differentially accessible regions using edgeR. As we do
not observe strong effects of GC content on signal neither in peaks nor
in genomic bins, we decided to use the scaling normalisation by trimmed
mean of M-values (TMM) (Robinson and Oshlack 2010).
We start by creating DGEList, the object edgeR uses to store
data for calculations. Before we start the DA analysis, it’s advisable
to remove peaks with very low counts.
reads.dge = DGEList(counts=reads.peak, group=groups)
keep = filterByExpr(reads.dge)
reads.dge=reads.dge[keep,,keep.lib.sizes=FALSE]
summary(keep)
## Mode FALSE TRUE
## logical 418 64461
reads.dge
## An object of class "DGEList"
## $counts
## B1_WT_Batf-floxed B2_WT_Batf-floxed A1_Batf_cKO A2_Batf_cKO
## merged_peaks_1 299 238 325 330
## merged_peaks_2 106 83 162 174
## merged_peaks_3 19 24 25 21
## merged_peaks_4 27 31 40 29
## merged_peaks_5 114 101 65 151
## 64456 more rows ...
##
## $samples
## group lib.size norm.factors
## B1_WT_Batf-floxed ctrl 43359738 1
## B2_WT_Batf-floxed ctrl 33327965 1
## A1_Batf_cKO KO_Batf 43438468 1
## A2_Batf_cKO KO_Batf 46400831 1
These steps perform the standard edgeR workflow for differential
analysis:
reads.dge.tmm = normLibSizes(reads.dge)
We can inspect sample grouping on multidimensional scaling (MDS) plot before proceeding:
plotMDS(reads.dge.tmm)

All looks as expected, we can proceed with the differential analysis:
reads.dge.tmm = estimateDisp(reads.dge.tmm, design)
qlf.fit.tmm=glmQLFit(reads.dge.tmm, design, robust=TRUE)
qlf.ftest.tmm=glmQLFTest(qlf.fit.tmm, coef=2)
DA_res.qlf.tmm=as.data.frame(topTags(qlf.ftest.tmm, nrow(qlf.ftest.tmm$table)))
DA_res.qlf.tmm=DA_res.qlf.tmm|>dplyr::mutate(peakID=rownames(DA_res.qlf.tmm))
This results in a table with results of DA analysis:
head(DA_res.qlf.tmm)
## logFC logCPM F PValue FDR
## merged_peaks_28038 -1.610768 6.074221 756.5472 1.722979e-90 1.110650e-85
## merged_peaks_51767 -1.508490 6.159745 710.5749 3.363346e-87 1.084023e-82
## merged_peaks_2997 -1.517878 5.964106 638.5088 9.578557e-82 2.058145e-77
## merged_peaks_1873 -1.157643 6.593339 534.2112 4.151681e-73 6.690538e-69
## merged_peaks_36974 -1.141022 6.596217 524.9430 2.716748e-72 3.502486e-68
## merged_peaks_40709 -1.638902 5.206780 468.3368 4.081673e-67 4.385146e-63
## peakID
## merged_peaks_28038 merged_peaks_28038
## merged_peaks_51767 merged_peaks_51767
## merged_peaks_2997 merged_peaks_2997
## merged_peaks_1873 merged_peaks_1873
## merged_peaks_36974 merged_peaks_36974
## merged_peaks_40709 merged_peaks_40709
We should also take a look at the diagnostic plots to verify that they look as expected. The MA plot is a visualisation that plots the log-fold-change between experimental groups (M) against the average expression across all the samples (A) for each gene. The expectation is that for the bulk of the peaks the log2FC is close to 0 (the cloud of points is centred on 0 on the Y-axis) and minimal signal amplitude related effects.
plotMD(qlf.ftest.tmm)

At this point we can add the information from peak annotation Peak Annotation
If you are still in the same R session, you can skip the step below.
If you started a new R session, you can read in the table with peak annotations:
peak_annots_pth=file.path(workdir,"Allpeaks_annot.Ensembl.rds")
peakAnno_df=readRDS(peak_annots_pth)
#rename the column with peakID for data frame joining and remove redundant columns
peakAnno_df=peakAnno_df|>dplyr::rename(peakID=mcols.peakID)|>dplyr::select(!c(seqnames,strand.x,start,end,width,strand.y))
Note
If you did not follow the Peak Annotation lab, you copy the saved file from
../../results/DA/objects/Allpeaks_annot.Ensembl.rds
We can now join the tables with peak annotations and DA results:
peaks_gr_df=as.data.frame(peaks_gr)|>dplyr::rename(peakID=mcols.peakID)
DA_res_table=DA_res.qlf.tmm |>
dplyr::left_join(peakAnno_df,by="peakID")|>
dplyr::left_join(peaks_gr_df,by="peakID")|>
dplyr::select(seqnames,start,end,peakID,gc,logFC,FDR,annotation,geneChr,geneStart,geneEnd,geneStrand,geneId,transcriptId,external_gene_name,distanceToTSS)
head(DA_res_table)
## seqnames start end peakID logFC FDR
## 1 17 66268427 66269247 merged_peaks_28038 -1.610768 1.110650e-85
## 2 6 122504236 122505014 merged_peaks_51767 -1.508490 1.084023e-82
## 3 1 155076669 155077704 merged_peaks_2997 -1.517878 2.058145e-77
## 4 1 95195320 95196614 merged_peaks_1873 -1.157643 6.690538e-69
## 5 2 162944874 162945676 merged_peaks_36974 -1.141022 3.502486e-68
## 6 3 138125917 138126743 merged_peaks_40709 -1.638902 4.385146e-63
## gc annotation
## 1 0.4360536 Distal Intergenic
## 2 0.4801027 Intron (ENSMUST00000032210/ENSMUSG00000030116, intron 8 of 8)
## 3 0.5009653 3' UTR
## 4 0.4617761 Distal Intergenic
## 5 0.5031133 Distal Intergenic
## 6 0.4087062 Exon (ENSMUST00000161312/ENSMUSG00000037797, exon 4 of 6)
## geneChr geneStart geneEnd geneStrand geneId transcriptId
## 1 17 66261129 66265392 1 ENSMUSG00000139744 ENSMUST00000355127
## 2 6 122499458 122505594 1 ENSMUSG00000030116 ENSMUST00000126357
## 3 1 155070767 155077993 1 ENSMUSG00000026470 ENSMUST00000194158
## 4 1 95183688 95184535 2 ENSMUSG00000099592 ENSMUST00000190584
## 5 2 162934819 162934943 1 ENSMUSG00002076785 ENSMUST00020181897
## 6 3 138121256 138136653 1 ENSMUSG00000037797 ENSMUST00000013458
## external_gene_name distanceToTSS
## 1 Gm65735 7298
## 2 Mfap5 4778
## 3 Stx6 5902
## 4 Gm5264 -10785
## 5 Gm56299 10055
## 6 Adh4 4661
We can save the complete results of peak annotation, GC content and DA analysis:
saveRDS(DA_res_table, file = "FiltPeaks.DA.TMM.annot.rds")
You can now follow with other downstream tutorials listed under Downstream Analyses
GC Bias Correction
Plotting Log2FC in GC Bins
When a strong effect of GC content on signal is observed, a GC aware scaling normalisation can be considered. It is important to perform all diagnostic plots, however, to verify whether it does not distort the data in an unexpected manner. One should always be aware that the GC bias, although technical, may also reflect sample biology, therefore removing it may lead to signal loss.
We can first verify whether there is a GC bias in log2FC detection using GC agnostic TMM scaling.
We can plot log2FC distribution in GC content bins.
For this we will need the GC bins we calculated before, so we need to join that information to the results of DA analysis:
peak_info_df=as.data.frame(peaks_gr)|>
dplyr::rename(peakID=mcols.peakID)
df_GCbias=DA_res.qlf.tmm |>
dplyr::left_join(peak_info_df, by="peakID") |>
dplyr::select(logFC,gc_group)
Let’s plot the log2FC in GC bins:
plot_lfc_GC_TMM = ggplot(df_GCbias) +
aes(x=gc_group, y=logFC, color=gc_group) +
geom_violin(width=0.95) +
geom_boxplot(width=0.15, color="grey20") +
scale_color_manual(values=wesanderson::wes_palette("Zissou1", nlevels(df_GCbias$gc_group), "continuous")) +
geom_abline(intercept = 0, slope = 0, col="black", lty=2) +
#ylim(c(-1,1)) + ## this was in the original code from EDAseq paper; it calculates medians for values within the ylim interval - not from the entire data
coord_cartesian(ylim=c(-1,1)) +
ggtitle(paste0("log2FCs in bins by GC content, normalisation: TMM")) +
xlab("GC-content bin") +
theme_bw()+
theme(axis.text.x = element_text(angle = 45, vjust = .5),
legend.position = "none",
axis.title = element_text(size=16))
plot_lfc_GC_TMM

A negligible bias in log2FC can be observed within the range of log2FC (-1,1).
You can alter the plotted range by changing
coord_cartesian(ylim=c(-1,1)) to your desired range.
In any case, for this data set the systematic effect of GC contents on detected log2FC is very small, and below the reasonable size effect cutoff.
Correcting for GC contents
If required, the raw counts can be scaled in a GC aware manner, rather than using the TMM method.
Two related methods are presented below. Both perform conditional
quantile scaling, and output the offsets which can then be used in
edgeR statistical framework.
Full Quantile GC-GC Scaling
This method is implemented in Bioconductor package EDASeq (Risso et
al. 2011).
To calculate the offsets, which correct for library size as well as GC content (full quantile normalisation in both cases):
exprsSet.eda=newSeqExpressionSet(reads.dge$counts)
peaks_gr.keep=peaks_gr[keep]
fData(exprsSet.eda)$gc=peaks_gr.keep$gc
exprsSet.eda.wl=withinLaneNormalization(exprsSet.eda,"gc",num.bins=20, which="full",offset=TRUE)
exprsSet.eda.bl=betweenLaneNormalization(exprsSet.eda.wl,which="full",offset=TRUE)
The offsets can be inspected:
head(offst(exprsSet.eda.bl))
## B1_WT_Batf-floxed B2_WT_Batf-floxed A1_Batf_cKO A2_Batf_cKO
## merged_peaks_1 1.2280870 1.5061165 1.2441529 1.1811897
## merged_peaks_2 1.0093249 1.2584039 1.1587582 1.0848913
## merged_peaks_3 0.6129416 0.9425809 0.7350182 0.5523253
## merged_peaks_4 0.7139578 0.9909705 0.8238461 0.6286266
## merged_peaks_5 0.6563096 0.9560009 0.5779452 0.6904069
## merged_peaks_6 0.6932922 1.0369034 0.7632731 0.7825120
We will input the offset matrix to edgeR:
reads.dge.edaseq = reads.dge
reads.dge.edaseq$offset = -offst(exprsSet.eda.bl)
The statistical testing follows:
reads.dge.edaseq=estimateDisp(reads.dge.edaseq, design)
qlf.fit.edaseq=glmQLFit(reads.dge.edaseq, design, robust=TRUE)
qlf.ftest.edaseq=glmQLFTest(qlf.fit.edaseq, coef=2)
DA_res.qlf.edaseq=as.data.frame(topTags(qlf.ftest.edaseq, nrow(qlf.ftest.edaseq$table)))
DA_res.qlf.edaseq=DA_res.qlf.edaseq|>dplyr::mutate(peakID=rownames(DA_res.qlf.edaseq))
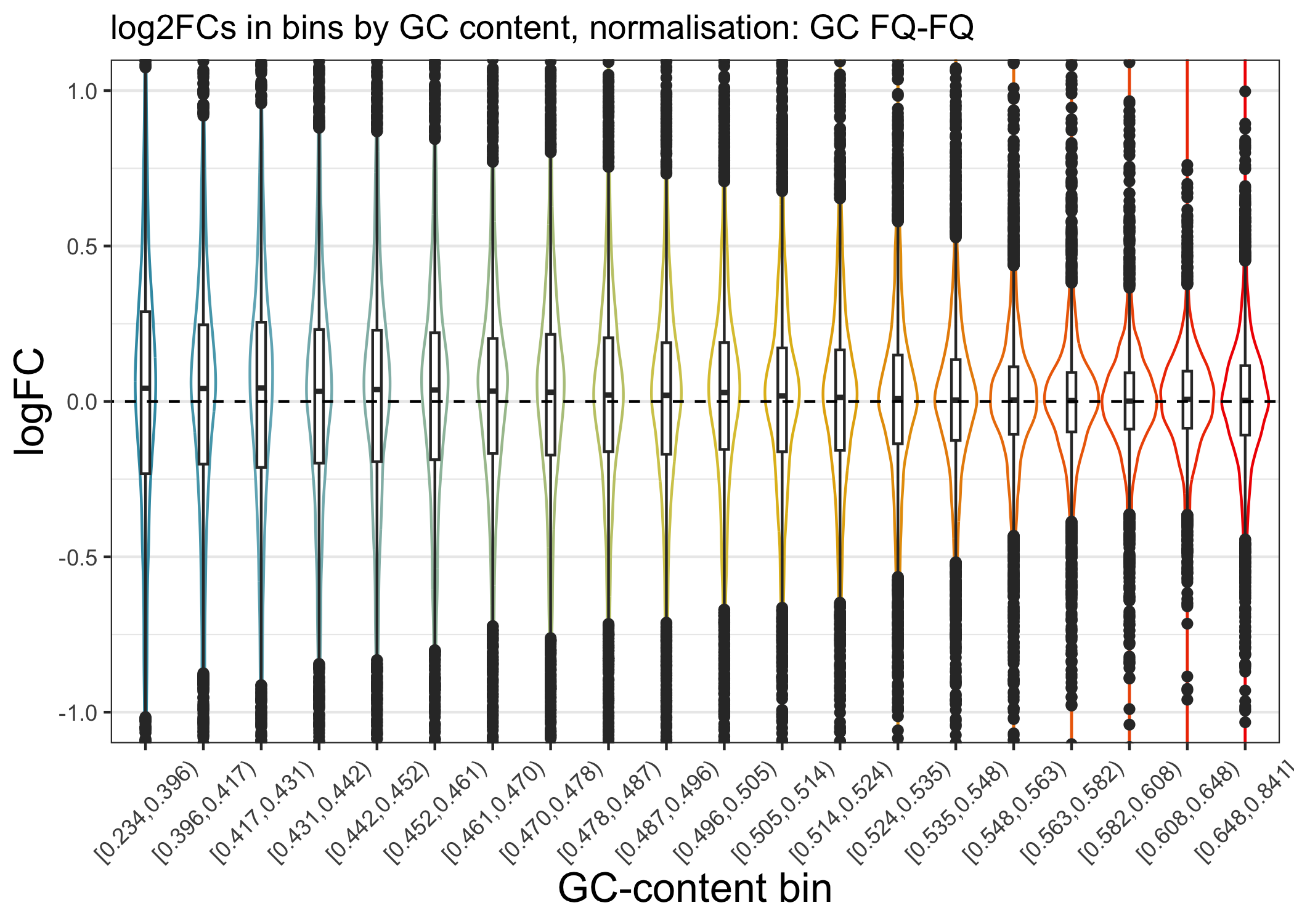
We can now plot the log2FC in GC bins, as for TMM scaling:
df_GCbias=DA_res.qlf.edaseq |>
dplyr::left_join(peak_info_df, by="peakID") |>
dplyr::select(logFC,gc_group)
plot_lfc_GC_edaseq = ggplot(df_GCbias) +
aes(x=gc_group, y=logFC, color=gc_group) +
geom_violin(width=0.95) +
geom_boxplot(width=0.15, color="grey20") +
scale_color_manual(values=wesanderson::wes_palette("Zissou1", nlevels(df_GCbias$gc_group), "continuous")) +
geom_abline(intercept = 0, slope = 0, col="black", lty=2) +
#ylim(c(-1,1)) + ## this was in the original code from EDAseq paper; it calculates medians for values within the ylim interval - not from the entire data
coord_cartesian(ylim=c(-1,1)) +
ggtitle(paste0("log2FCs in bins by GC content, normalisation: GC FQ-FQ")) +
xlab("GC-content bin") +
theme_bw()+
theme(axis.text.x = element_text(angle = 45, vjust = .5),
legend.position = "none",
axis.title = element_text(size=16))
plot_lfc_GC_edaseq
Conditional Quantile Normalization
This method is implemented in Bioconductor package cqn (Hansen,
Irizarry, and WU 2012).
In calculating offsets, it can correct both for GC content as well as peak length.
#assuming we have the subset peaks_gr
peaks_gr.keep=peaks_gr[keep]
peaks=as.data.frame(cbind(
gc=peaks_gr.keep$gc,
length=width(peaks_gr.keep)
))
rownames(peaks)=peaks_gr.keep$mcols.peakID
cqn_out=cqn(counts=reads.dge$counts,lengths=peaks$length,x=peaks$gc,
sizeFactors=reads.dge$samples$lib.size,verbose=TRUE)
## RQ fit ....
## SQN
## .
cqn_out
##
## Call:
## cqn(counts = reads.dge$counts, x = peaks$gc, lengths = peaks$length,
## sizeFactors = reads.dge$samples$lib.size, verbose = TRUE)
##
## Object of class 'cqn' with
## 64461 regions
## 4 samples
## fitted using smooth length
head(cqn_out$glm.offset)
## B1_WT_Batf-floxed B2_WT_Batf-floxed A1_Batf_cKO A2_Batf_cKO
## merged_peaks_1 6.077630 5.849091 6.038654 6.142638
## merged_peaks_2 5.602194 5.392023 5.695700 5.805515
## merged_peaks_3 3.810420 3.669992 3.829517 3.872245
## merged_peaks_4 3.460506 3.172147 3.333104 3.457118
## merged_peaks_5 5.812975 5.660115 5.655834 6.000880
## merged_peaks_6 5.993356 5.874478 5.930493 6.225818
We will input the offset matrix to edgeR:
reads.dge.cqn = reads.dge
reads.dge.cqn$offset = cqn_out$glm.offset
The statistical testing follows:
reads.dge.cqn=estimateDisp(reads.dge.cqn, design)
qlf.fit.cqn=glmQLFit(reads.dge.cqn, design, robust=TRUE)
qlf.ftest.cqn=glmQLFTest(qlf.fit.cqn, coef=2)
DA_res.qlf.cqn=as.data.frame(topTags(qlf.ftest.cqn, nrow(qlf.ftest.cqn$table)))
DA_res.qlf.cqn=DA_res.qlf.cqn|>dplyr::mutate(peakID=rownames(DA_res.qlf.cqn))
We can now plot the log2FC in GC bins, as for TMM scaling:
df_GCbias=DA_res.qlf.cqn |>
dplyr::left_join(peak_info_df, by="peakID") |>
dplyr::select(logFC,gc_group)
plot_lfc_GC_cqn = ggplot(df_GCbias) +
aes(x=gc_group, y=logFC, color=gc_group) +
geom_violin(width=0.95) +
geom_boxplot(width=0.15, color="grey20") +
scale_color_manual(values=wesanderson::wes_palette("Zissou1", nlevels(df_GCbias$gc_group), "continuous")) +
geom_abline(intercept = 0, slope = 0, col="black", lty=2) +
#ylim(c(-1,1)) + ## this was in the original code from EDAseq paper; it calculates medians for values within the ylim interval - not from the entire data
coord_cartesian(ylim=c(-1,1)) +
ggtitle(paste0("log2FCs in bins by GC content, normalisation: cqn")) +
xlab("GC-content bin") +
theme_bw()+
theme(axis.text.x = element_text(angle = 45, vjust = .5),
legend.position = "none",
axis.title = element_text(size=16))
plot_lfc_GC_cqn
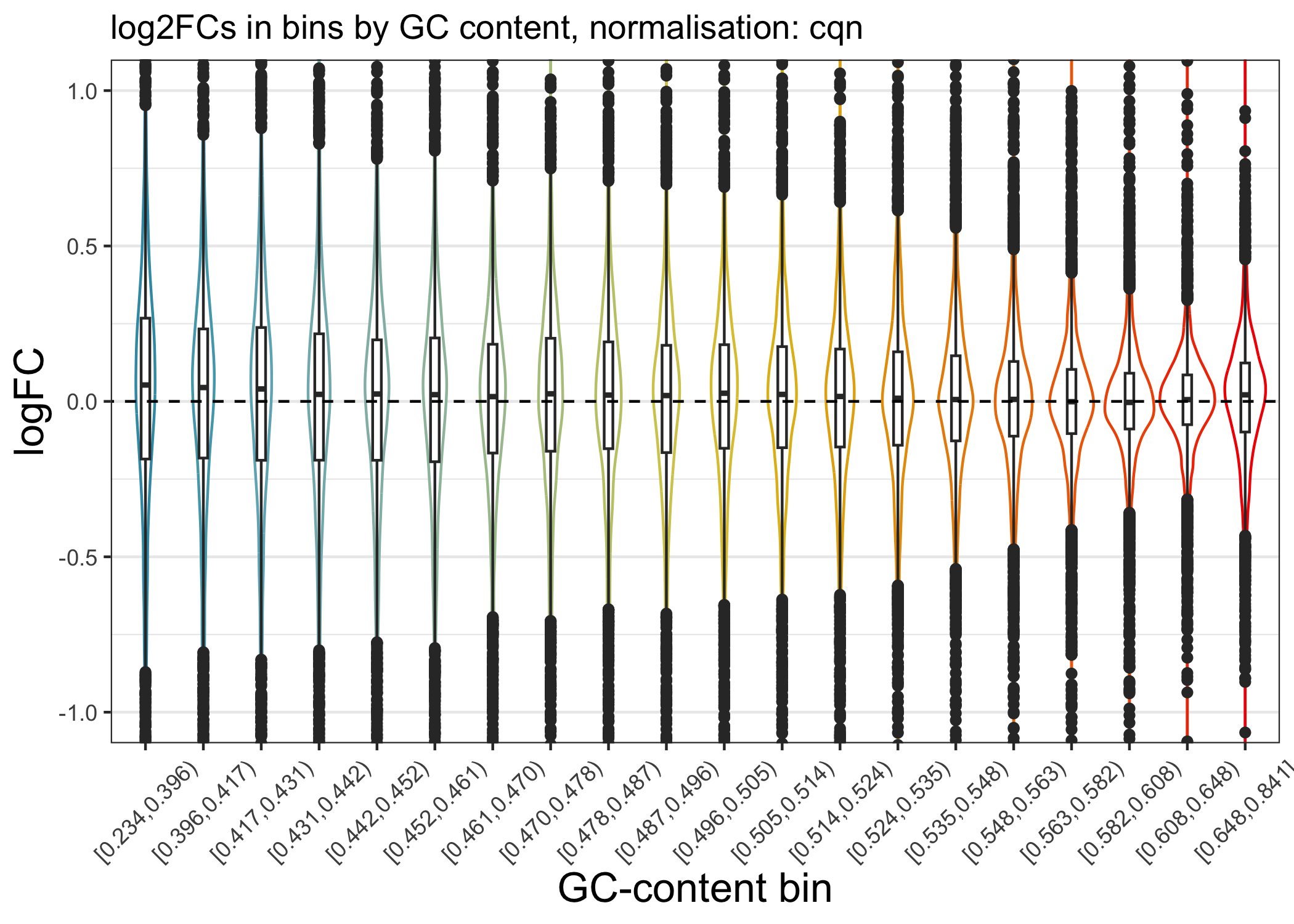
Note
It is advised to verify the estimated model parameters and fit using the
diagnostic plots provided in edgeR i.e. plotBCV(reads.dge) and
plotQLDisp(fit)
Session Info
Session Info.
## R version 4.4.2 (2024-10-31)
## Platform: x86_64-apple-darwin20
## Running under: macOS Sonoma 14.5
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_GB.UTF-8
##
## time zone: Europe/Stockholm
## tzcode source: internal
##
## attached base packages:
## [1] splines stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] cqn_1.50.0 quantreg_6.1
## [3] SparseM_1.84-2 preprocessCore_1.66.0
## [5] nor1mix_1.3-3 mclust_6.1.1
## [7] EDASeq_2.38.0 ShortRead_1.62.0
## [9] GenomicAlignments_1.40.0 Rsamtools_2.20.0
## [11] BiocParallel_1.38.0 SummarizedExperiment_1.34.0
## [13] Biobase_2.64.0 MatrixGenerics_1.16.0
## [15] matrixStats_1.5.0 edgeR_4.2.2
## [17] limma_3.60.6 BSgenome.Mmusculus.UCSC.mm39_1.4.3
## [19] BSgenome_1.72.0 rtracklayer_1.64.0
## [21] BiocIO_1.14.0 MASS_7.3-65
## [23] bamsignals_1.36.0 regioneR_1.36.0
## [25] Biostrings_2.72.1 XVector_0.44.0
## [27] Hmisc_5.2-3 GenomicRanges_1.56.2
## [29] GenomeInfoDb_1.40.1 IRanges_2.38.1
## [31] S4Vectors_0.42.1 BiocGenerics_0.50.0
## [33] wesanderson_0.3.7 kableExtra_1.4.0
## [35] lubridate_1.9.4 forcats_1.0.0
## [37] stringr_1.5.2 dplyr_1.1.4
## [39] purrr_1.1.0 readr_2.1.5
## [41] tidyr_1.3.1 tibble_3.3.0
## [43] ggplot2_3.5.2 tidyverse_2.0.0
## [45] bookdown_0.44 knitr_1.50
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 rstudioapi_0.17.1 jsonlite_2.0.0
## [4] magrittr_2.0.3 GenomicFeatures_1.56.0 farver_2.1.2
## [7] rmarkdown_2.29 zlibbioc_1.50.0 vctrs_0.6.5
## [10] memoise_2.0.1 RCurl_1.98-1.17 base64enc_0.1-3
## [13] progress_1.2.3 htmltools_0.5.8.1 S4Arrays_1.4.1
## [16] curl_7.0.0 SparseArray_1.4.8 Formula_1.2-5
## [19] KernSmooth_2.23-26 htmlwidgets_1.6.4 httr2_1.2.1
## [22] cachem_1.1.0 lifecycle_1.0.4 pkgconfig_2.0.3
## [25] Matrix_1.7-4 R6_2.6.1 fastmap_1.2.0
## [28] GenomeInfoDbData_1.2.12 digest_0.6.37 colorspace_2.1-1
## [31] AnnotationDbi_1.66.0 textshaping_1.0.3 RSQLite_2.4.3
## [34] hwriter_1.3.2.1 labeling_0.4.3 filelock_1.0.3
## [37] timechange_0.3.0 httr_1.4.7 abind_1.4-8
## [40] compiler_4.4.2 bit64_4.6.0-1 withr_3.0.2
## [43] htmlTable_2.4.3 backports_1.5.0 DBI_1.2.3
## [46] hexbin_1.28.5 R.utils_2.13.0 biomaRt_2.60.1
## [49] rappdirs_0.3.3 DelayedArray_0.30.1 rjson_0.2.23
## [52] tools_4.4.2 foreign_0.8-90 nnet_7.3-20
## [55] R.oo_1.27.1 glue_1.8.0 restfulr_0.0.16
## [58] grid_4.4.2 checkmate_2.3.3 cluster_2.1.8.1
## [61] generics_0.1.4 gtable_0.3.6 tzdb_0.5.0
## [64] R.methodsS3_1.8.2 data.table_1.17.8 hms_1.1.3
## [67] xml2_1.4.0 pillar_1.11.0 BiocFileCache_2.12.0
## [70] lattice_0.22-7 survival_3.8-3 aroma.light_3.34.0
## [73] bit_4.6.0 deldir_2.0-4 tidyselect_1.2.1
## [76] locfit_1.5-9.12 gridExtra_2.3 svglite_2.2.1
## [79] xfun_0.53 statmod_1.5.0 stringi_1.8.7
## [82] UCSC.utils_1.0.0 yaml_2.3.10 evaluate_1.0.5
## [85] codetools_0.2-20 interp_1.1-6 cli_3.6.5
## [88] rpart_4.1.24 systemfonts_1.2.3 Rcpp_1.1.0
## [91] dbplyr_2.5.0 png_0.1-8 XML_3.99-0.19
## [94] parallel_4.4.2 MatrixModels_0.5-4 blob_1.2.4
## [97] prettyunits_1.2.0 latticeExtra_0.6-30 jpeg_0.1-11
## [100] bitops_1.0-9 pwalign_1.0.0 viridisLite_0.4.2
## [103] scales_1.4.0 crayon_1.5.3 rlang_1.1.6
## [106] KEGGREST_1.44.1
References
Chen, Yunshun, Aaron T. L. Lun, and Gordon K. Smyth. 2016. “From Reads to Genes to Pathways: Differential Expression Analysis of RNA-Seq Experiments Using Rsubread and the edgeR Quasi-Likelihood Pipeline.” F1000Research 5 (August): 1438. https://doi.org/10.12688/f1000research.8987.2.
Hansen, K. D., R. A. Irizarry, and Z. WU. 2012. “Removing Technical Variability in RNA-Seq Data Using Conditional Quantile Normalization.” Biostatistics 13 (2): 204–16. https://doi.org/10.1093/biostatistics/kxr054.
Risso, Davide, Katja Schwartz, Gavin Sherlock, and Sandrine Dudoit. 2011. “GC-Content Normalization for RNA-Seq Data.” BMC Bioinformatics 12 (1). https://doi.org/10.1186/1471-2105-12-480.
Robinson, M. D., and A. Oshlack. 2010. “A scaling normalization method for differential expression analysis of RNA-seq data.” Genome Biol 11 (3): R25.
Tsao, Hsiao-Wei, James Kaminski, Makoto Kurachi, R. Anthony Barnitz, Michael A. DiIorio, Martin W. LaFleur, Wataru Ise, et al. 2022. “Batf-Mediated Epigenetic Control of Effector CD8 + t Cell Differentiation.” Science Immunology 7 (68). https://doi.org/10.1126/sciimmunol.abi4919.