Experimental Design Considerations for ChIP-seq
Contents:
Note
Sound experimental design includes replication, randomisation, control and blocking (R.A. Fisher, 1935).
In the absence of a proper design, it is essentially impossible to partition biological variation from technical variation.
General Comments
There are several factors to consider when planning a ChIP-seq experiment:
Sequencing depth: depends on the structure of the signal; cannot be linearly scaled to genome size;
Single- vs. paired-end reads: PE improves read mapping confidence and gives a direct measure of fragment size, which otherwise has to be modelled or estimated;
Other factors: cross-linker choice, chromatin fragmentation method, antibody, …
Below we attempted to summarise the most important issues to consider when desiging a ChIP-seq experiment. Effects of some common pitfalls are illustarted.
Biological replicates are a must in any experiment. Three is a minimum for statistical analysis of occupancy patterns between differnet conditions, two may be enough for descriptive binding characterisation, but often it is recommended to sequence more replicates if possible (Yang 2014). It’s worth to remember that if small differences in occupancy are expected, the number of replicates brings more to the table than deeper sequencing. Sequencing of technical replicates is not necessary.
Controls are crucial for analysis of ChIP-seq data. They are used to model the local structure of ChIP-seq signal, and it is impossible to detect relaiable binding events (peaks) without them. Two types of controls are used for ChIP-seq experiments, input chromatin and IgG IP. Input chromatin is more widely used, and it seems to be less biased than IgG, which also often suffers from library complexity issues (Marinov 2014).
ChIP-seq experiment design, replicates
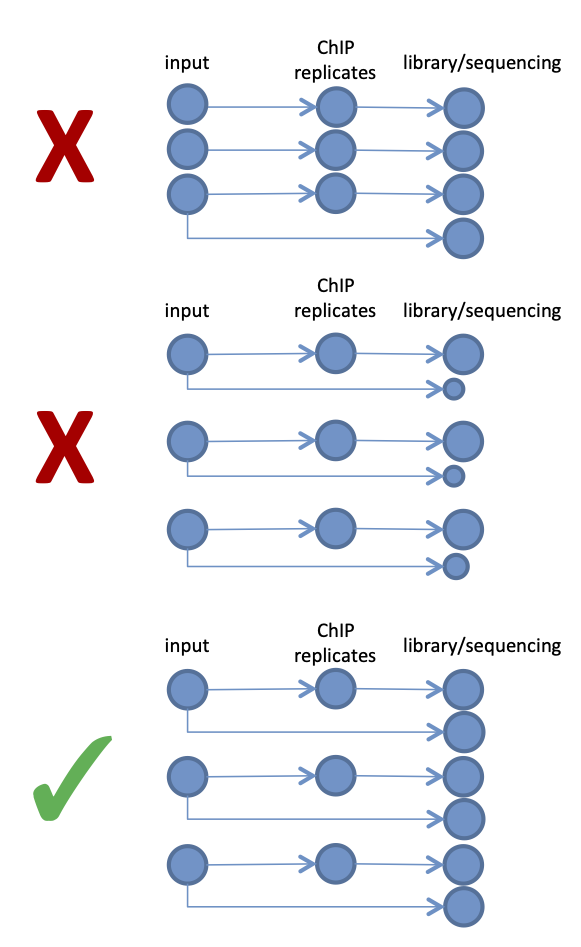
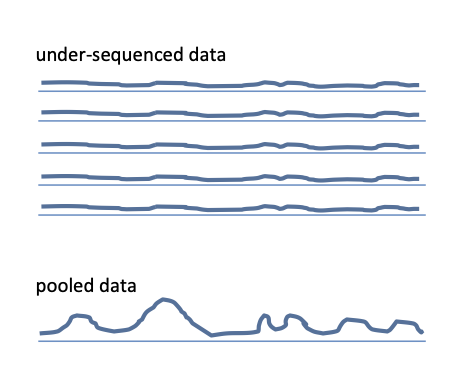
Input control (chromatin or IgG) should be sequenced to at least the same depth than the ChIP samples. Each replicate of ChIP should have its own matching input which should be sequenced separately from other input samples (i.e. no pooling of inputs). Only then the local fluctuations in the background signal can be properly assesed.
ChIP-seq experiment design, input sequencing depth
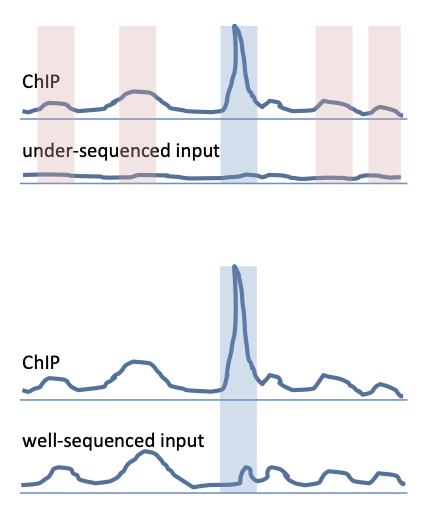
Recommended sequencing depth for several types of signal is listed in Table 1. Recommended depth (library size) refers to uniquely mapped reads i.e. reads (SE) or fragments (PE) with one best match for human data. These recommendations are probably valid for other mamammalian data; for other organisms please see Jung et al, NAR 2014.
Signal |
Example |
Recommended depth |
|---|---|---|
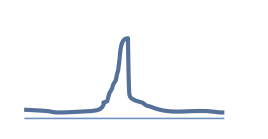
|
Point source: transcription factors, H3K4me3 |
20 - 25 M |
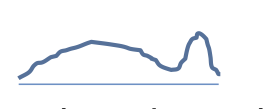
|
Mixed: H3K36me3 |
35M |
|
Broad signal: chromatin remodellers, histone marks, RNA polymerase II |
H3K27me3: 40M; H3K9me3 > 55M? |
Source: The ENCODE consortium guidelines; Jung et al 2014.
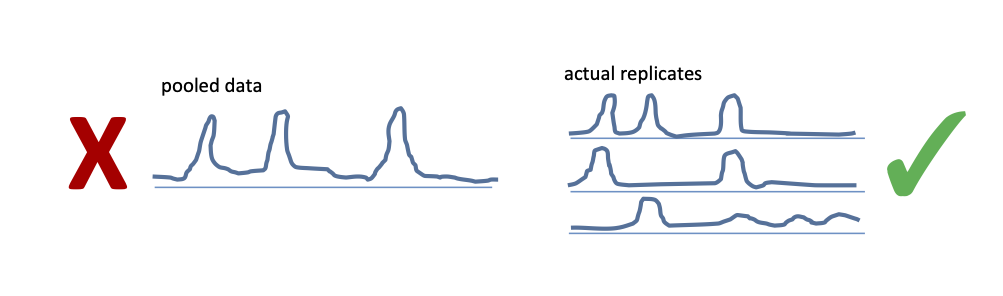
It is vital that the samples are sequenced to depth sufficient to detect binding events in each replicate independently. If the replicates must be pooled to detect peaks, the sequencig was too shallow.
ChIP-seq experiment design, find peaks in each sample
ChIP-seq experiment design, sample sequencing depth
PE vs. SE is usually a question of cost. If working with a factor with point source occupancy pattern, SE is usually enough. However, investigating broader occupancy patterns does benefit from PE data, as fragment length does not need to be modelled (which is almost never accurate for this type of data), and is taken directly from data.
When interpreting quality of your experiment, we recommend to refer to The ENCODE consortium guidelines.
We recommend performing a pilot experiment with small number of samples if unsure whether selected design will deliver data you need to answer your biological question.
Considerations for Point Source Factors
Transcription Factors, H3K4me3
20 - 25 M reads, may be SE.
Considerations for Broad Enrichment Domains
Histone modifications, chromatin remodellers, RNA polymerases
There are no clear guidelines as to sequencing depth for mixed and broad type of peaks. The numbers in Table 1 are only estimates. ENCODE recommends 45 M for broad marks (H3K27me3, H3K36me3, H3K4me1, etc) (ENCODE histone ChIPseq guidelines). PE is recommended.
A pilot experiment may be necessary if working with non-histone factor, whose occupancy has not been well characterised.